NEW STUDY DISPROVES ALL MAINSTREAM THEORIES OF AGING
AND REVEALS THE NEW:
PROGRAMMED LOSS
OF
CELLULAR DIFFERENTIATION
THEORY OF AGING
AND
A HUGE NEW PROBLEM FOR EVOUTIONARY BIOLOGISTS
(ACTUALLY THERE ALSO CO-EXISTS A SECOND SMALLER AGING SYSTEM THAT INVOLVES THE ACTIVATION OF PRO-AGING GENES BY AGE-RELATED CHANGES IN HORMONES-BUT THIS IS OLDER NEWS-AT LEAST TO ME & IT WILL ALSO BE DISCUSSED HEREIN)
Keep checking back as this post is often updated! Updates usually added at the end.
MAKE SURE YOU CHECK OUT UPDATE #9 – It is amazing! #13 is pretty cool too!
See Update #23 For Another study that proves all of what you are about to read is correct- aging was recently reversed in old rats by 50 to 75% using the ideas discussed herein!
Update #24 7/19/2021 Update #25 8/5/2021 Aging reversed by 2 years in humans with GH, DHEA, and metformin
Feeling lazy?>>> watch these podcasts >>>. https://youtu.be/61OagronJ64
or >>>>>>>>> https://youtu.be/UsQ3D8BaYVk but make sure you look over this blog post afterwards has a lot more interesting explanations of additional very interesting things like progeria and Werner’s syndrome , rapidly aging salmon, etc.
NEW PUBMED search tool….. search all 48 Horvath aging genes against your topic of interest (For example- WRN or telomere) and see which genes pop up…..at the end of this post…let me know if you find anything exciting!
JEFF T. BOWLES
11/22/2022
Before we get started let me just whet your appetite about what is contained in the rest of this article. The results of the most important study on aging EVER, that will be the most important study of aging for all time- have just been released! Steve Horvath’s :
Universal DNA methylation age across mammalian tissues
The study proves conclusively that aging is selected for by evolution and is programmed. A result that contradicts all major mainstream theories of aging that have been proposed since the early 1900’s. It turns out August Weisman got the right answer in 1882 but with the wrong reasoning.
The new study also reveals the true cause of aging at the cellular level- the programmed loss of cellular differentiation.
Recently, a preprint of a journal article that is expected to be published in Nature, was released that completely breaks open the cause of aging in mammals of almost all species. The paper shows that this aging is highly conserved by evolution and ends the debate about whether aging is caused by accidental DNA damage or is programmed. The answer?- aging evolved, is highly conserved, and is programmed- no doubt about it!
The paper was lead-authored by Steve Horvath and co-authored with a long list of collaborators. It is currently titled>> Universal DNA methylation age across mammalian tissues.
I think it is the most important study concerning aging and always will be. And I have been studying aging for 35+ years and have seen almost everything! Horvath’s travels through the methylation of the DNA of so many animals is certainly as important as and probably more so than Darwin’s 5 years on the HMS Beagle.
Here is the link to the preprint>> https://www.biorxiv.org/content/10.1101/2021.01.18.426733v1.full
In the last sentence of his abstract Horvath bravely states-
“Collectively, these new observations support the notion that aging is indeed evolutionarily conserved and coupled to developmental processes across all mammalian species – a notion that was long-debated without the benefit of this new and compelling evidence.”
NOT ONLY IS THIS THE MOST IMPORTANT STUDY CONCERNING AGING EVER- IT ALSO PROVES
THAT
THE MAINSTREAM SCIENCE’S VERSION OF THE THEORY OF EVOLUTION IS WRONG!
All those evolution professors are going to have to go back to the drawing board because in their view of evolution it is impossible for aging to have evolved and be selected for because it is bad for the spread of your selfish genes! Virtually all evolutionary biologists believe that aging being selected for by evolution is impossible!
Actually, I propose in another article with a link at the end of this one, that the selfish gene theory of evolution is mostly correct but it is only half the story. There is a missing half of the theory of evolution that has yet to be revealed. I take a stab at it, and succeed, in the article linked to at the end. Okay back to aging…
Here is a summary of what Horvath et al found:
Horvath and this team looked at the DNA of a large number of mammals and determined what were the genes that experienced major changes of DNA methylation (both increases and decreases) at older ages. Increased methylation at the beginning of a gene would basically shut it down, removal of methylation from the beginning of a gene would allow that gene to be expressed at older ages
They looked at the DNA methylation changes with age in 59 different tissue types from 128 mammalian species to see what they all had in common.
They found a highly conserved aging program driven by DNA methylation changes that for the most part shut down genes that produced transcription factors by adding methyl groups to the promoter area of the genes. They found 36 genes that were affected /shut down by DNA methylation and almost all of them were transcription factors that are involved in the differentiation of cells during development that have homeobox domains. They found very few (12) genes that experienced loss of methylation which was a surprise to me based on my predictions in my 1998 paper . I expected it to be the other way around because the entire genome loses a lot of methylation during aging. So, these instances of hypermethylation must be very special to buck the overall trend in the global DNA demethylation with age, apparently most DNA methylation is uninvolved with direct aging control.
Overall, they found 3,617 cases of hypermethylated cytosines in the DNA associated with aging and only 12 hypomethylated cytosines! This blew my mind.
Well, those 12 hypomethylated sites must be next to some very interesting genes! They analyzed these hypomethylated genes and found the #1 gene that was most hypomethylated in liver and #2 across all tissue types was the LARP1 gene. This gene being more expressed at older ages must be doing something very naughty! Let us take a look at the LARP 1 gene’ function as described by Wikipedia>>>>
Well, what do you know?? LARP1 has a unique region that binds to RNA transcripts! My guess is that this is the protein that is involved in truncating the Lamin A protein in normally aging cells, and likely it is truncating the WRN protein in normally aging cells (truncated WRN protein being found in normally aging and senescent cells has yet to be shown true-but I predict someday this will be found to be occurring).
(If this LARP1 info is boring to you now you can skip over it to get to the good stuff and come back later!)
LARP1
From Wikipedia, the free encyclopedia
La-related protein 1 (LARP1) is a 150 kDa protein that in humans is encoded by the LARP1 gene.[5][6][7] LARP1 is a novel target of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, a circuitry often hyperactivated in cancer which regulates cell growth and proliferation primarily through the regulation of protein synthesis.[8]
Function
LARP1 is the largest of a 7-member family of LARP proteins (others are: LARP1B, LARP3 (aka genuine La or SSB), LARP4A, LARP4B, LARP6 and LARP7).[9] All LARP proteins, including human LARPs, contain 2 conserved regions. The first conserved region shares homology with La proteins (called the La motif, see SSB) whereas the second conserved region (called the LA- motif) is restricted to LARP proteins. LARP1 and 1B also contain a conserved “DM15 region” within their C-terminus.[10] This region is unique and has been shown to be required for RNA-binding. Mouse Larp1 is expressed in dorsal root ganglia and spinal cord, as well as in developing organs characterized by epithelial–mesenchymal interactions.[6] Human LARP1 is present at low levels in normal, non-embryonic cells but is highly expressed in epithelial cancers (such as ovarian, colorectal, prostate, non-small cell lung, hepatocellular and cervical cancers).[11][12][13][14] Some studies have shown that high levels of LARP1 protein correlate with worse prognosis in cancer patients.[15][16]
LARP1 binds to and regulates the translation of terminal oligopyrimidine motif (TOP mRNAs) and can directly interact with the 5′ cap of mRNAs.[17][18] It has also been shown to interact with the 3′ end and coding regions (CDS) of other genes.[17] LARP1 protein colocalizes with stress granules and P-bodies,[19] which function in RNA storage and degradation. It has been suggested that LARP1 functions in P-bodies to attenuate the abundance of conserved Ras–MAPK mRNAs. The cluster of LARP1 homologs may function to control the expression of key developmental regulators.[19]
Several studies have demonstrated that LARP1 deficiency selectively affects the recruitment of TOP mRNAs to polysomes In some cancer cells, LARP1 deficiency reduces proliferation and activates apoptotic cell death.[13] Even though a decrease abundance of proteins encoded by TOP mRNAs has been reported in LARP1 silenced cells, some researchers believe that this can be explained simply by the reduced number of TOP mRNA transcripts in LARP1-deficient cells.
It turns out I predicted most of this long ago in 1998.
In 1998, I published a paper titled “The Evolution of Aging: A New Approach to an Old Problem of Biology”
in Medical Hypotheses Sep 1998.
This paper was the result of almost 10 years of non-stop 7 day a week, feverish research at the Northwestern Medical School library where I read everything I could find about aging. At the end of 10 years, I was like the first paleontologist who had uncovered the complete skeleton of a dinosaur but the bones were strewn about. I had identified almost all the relevant factors related to aging. It was time to put the bones together to see what the dinosaur looked like. Just like that first paleontologist’s attempt, my assembled dinosaur (aging theory) was mostly correct, but there were some bones placed in the wrong position.

Many good predictions came out of the paper which proved to be true such as:
-Aging is driven by the loss of DNA methylation of cytosines (actually driven by cytosine’s gain or loss of methylation (CH3’s)) also known as epigenetics. The next paper confirming this prediction did not come out until 2012 > Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell 2012 Jan 20;148(1-2):46-57. (written by some guy at Stanford- who did not mention my 1998 paper )
-Alzheimer’s and dementia would be found to be driven by the increase in Luteinizing Hormone that occurs after age 50 in both men and women. Confirmed in 2005 at the NIH>> Evidence for the role of gonadotropin hormones in the development of Alzheimer disease. Cell Mol Life Sci. 2005 Feb;62(3):293-8.
-Luteinizing Hormone and Follicle Stimulating Hormone would be found to play a central role in aging . Confirmed >> Data mining of human plasma proteins generates a multitude of highly predictive aging clocks that reflect different aspects of aging. October 2020 Aging Cell 19(1):e13256 ( the #1 and #2 proteins that increase the most in the aging cell are related to LH(#1) and FSH (#2).)
-The Hierarchy of programmed aging control was predicted to be
Hormone Changes>> Loss of Methylation >>> Expression of genes that cause aging.
This study proves this to be true, but to a lesser extent than I expected. What I did not expect was another hierarchy revealed by this study
Hormone Changes >>>> Gain of Methylation >>> Suppression of genes required for cellular differentiation.
-And one more little thing predicted in my 98 paper , that aging EVOLVED and is PROGRAMMED and is controlled by the same things that control development.
-The first 2 sentences in the abstract of my paper claimed that aging evolved and aging and development were intimately linked. This new study proves it to be 100% true.
“The evolution of aging: a new approach to an old problem of biology”
Bowles, JT Medical Hypotheses Sep 98
Abstract
“Most gerontologists believe aging did not evolve, is accidental, and is unrelated to development.
The opposite viewpoint is most likely correct.”
The problems with the paper were caused by my trying to put all the aging puzzle pieces together without enough information. For example, I imagined that the protein that is defective in Werner’s Syndrome (truncated) was generating excessive free radicals during the DNA unwinding process that catalyzed the demethylation of cytosines in the DNA. I thought this allowed pro-aging genes to be expressed, filling the body with destructive proteins. I received endless ridicule and derision from mainstream aging theorists who believed that the evolution of pro-aging genes was impossible. The new study shows that there are pro-aging genes, just not as many as I had imagined. It turns out that a lot of the programmed aging is caused by the suppression of genes that make transcription factors involved in maintaining cellular differentiation during and after development.
In reality what was happening was that the WRN helicase consists of 6 identical subunits which come together to form a helicase. The job of a helicase is to unwind and rewind the DNA. The single subunit also has another job as a transcription factor that binds to and silences some genes in stem cells to allow them to retain their differentiation and remain stem cells. But this was not known when I wrote the paper, so I gave it my best guess.
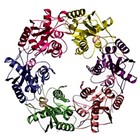
See>> A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging Science. 2015 Jun 5; 348(6239): 1160–1163.
Abstract
Werner syndrome (WS) is a premature aging disorder caused by WRN protein deficiency. Here, we report on the generation of a human WS model in human embryonic stem cells (ESCs). Differentiation of WRN-null ESCs to mesenchymal stem cells (MSCs) recapitulates features of premature cellular aging, a global loss of and changes in heterochromatin architecture. We show that WRN associates with heterochromatin proteins SUV39H1 and HP1α and nuclear lamina-heterochromatin anchoring protein LAP2β. Targeted knock-in of catalytically inactive SUV39H1 in wild-type MSCs recapitulates accelerated cellular senescence, resembling WRN-deficient MSCs. Moreover, decrease in WRN and heterochromatin marks are detected in MSCs from older individuals. Our observations uncover a role for WRN in maintaining heterochromatin stability and highlight heterochromatin disorganization as a potential determinant of human aging.
“Finally, we asked whether heterochromatin disorganization could be a common hallmark for physiological human stem cell aging. For this purpose, we compared the levels of heterochromatin marks in primary dental pulp MSCs derived from six young (7–26 year old) and six old (58–72 year old) individuals (fig. S10I, and Table S4) (20). A marked downregulation of WRN protein associated with a decrease in H3K9me3, HP1α, SUV39H1, and LAP2β levels in MSCs derived from old individuals (Fig. 4E). Therefore, specific heterochromatin changes may underlie both pathological as well as physiological mesenchymal stem cell aging.
In summary, we have found that WRN protein, besides its role in DNA repair, functions to safeguard heterochromatin stability (fig. S11). Our results unveil that the progressive heterochromatin disorganization observed in WRN deficient MSCs underlies cellular aging, but more extensive studies are needed to examine its role during physiological aging.”
Werner’s Syndrome is a rapid aging disease that kicks in around puberty and leads to thoroughly aged people by the age of 45 or so>>
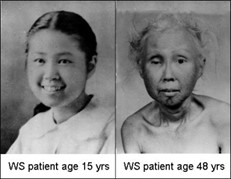

Werner’s Syndrome is caused by the WRN protein being improperly truncated so that it is too short to do its job properly of preserving the differentiation status of human stem cells.
Werner’s Syndrome is very much the same as normal aging. These patients have all the classic signs of aging , but they also have some extra-rare forms of disease which is what has led scientists to try and claim that this was not real aging. WS patients are afflicted with quite a few rare cancers as well as the normal aging processes.
I believe that the excess of rare cancers and other oddities associated with WS are caused not by the single truncated protein which causes all the features of normal aging, but rather by the improper functioning for the DNA helicase made by the 6 identical but defective WRN helicase subunits. Because proper functioning of DNA helicases are required for proper DNA maintenance and repair, it is not surprising that defective helicases would be associated with various odd forms of cancer.
(To my knowledge, truncated WRN protein being found in normally aging and senescent cells has yet to be discovered-but I predict someday this will be forthcoming).
The truncated differentiation/helicase protein found in Werner’s Syndrome is similar in concept to the disease called progeria which attacks young children from the time they are born and turns them into very old decrepit individuals by the age of 12 or so where they usually die of heart disease or atherosclerosis. Progeria is also caused by a truncated protein , the Lamin A protein which is a protein that is found in the nuclear envelope inside the cell. I proposed that progeria recapitulates many of the aging symptoms seen as more pronounced in normally aging males.


The truncated Lamin A protein causes the envelope that surrounds the DNA in the nucleus to be misshapen>>
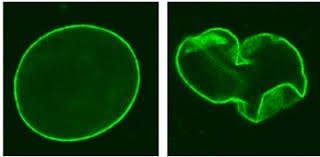 Normal Nucleus Progeria Nucleus
Normal Nucleus Progeria Nucleus
What is not that well known is that the progeria Lamin A protein has a 2nd function of binding to the DNA to act as a transcription factor that silences various genes so that various cells maintain their differentiation with the proper gene expression profile (for example so that a skin cell remains a skin cell by keeping a certain set of genes silenced).
See UPDATE #12 at the end of this article- it turns out that Lamin A is NOT expressed in induced pluripotent stem cells (undifferentiated cells) and the nucleus of these undifferentiated cells looks a lot like the nucleus in progeria cells! This gives further weight to the idea that aging is caused by loss of cellular differentiation.

Well, it turns out that this truncated Lamin A protein is not unique to progeria kids but is also seen in normal aging at older ages in normal adults! It is found in senescent cells in normal humans- there are a number of studies on this for example>>>
PLoS One. 2007; 2(12): e1269.
Published online 2007 Dec 5.
The Mutant Form of Lamin A that Causes Hutchinson-Gilford Progeria Is a Biomarker of Cellular Aging in Human Skin
Abstract Hutchinson-Gilford progeria syndrome is a rare disorder characterized by accelerated aging and early death, frequently from stroke or coronary artery disease. 90% of HGPS cases carry the LMNA G608G (GGC>GGT) mutation within exon 11 of LMNA, activating a splice donor site that results in production of a dominant negative form of lamin A protein, denoted progerin. Screening 150 skin biopsies from unaffected individuals (newborn to 97 years) showed that a similar splicing event occurs in vivo at a low level in the skin at all ages. While progerin mRNA remains low, the protein accumulates in the skin with age in a subset of dermal fibroblasts and in a few terminally differentiated keratinocytes. Progerin-positive fibroblasts localize near the basement membrane and in the papillary dermis of young adult skin; however, their numbers increase, and their distribution reaches the deep reticular dermis in elderly skin. Our findings demonstrate that progerin expression is a biomarker of normal cellular aging and may potentially be linked to terminal differentiation and senescence in elderly individuals. “
So, in both cases of Werner’s Syndrome and progeria we find a truncated protein that is used for differentiating cells is defective and unable to properly do its job of maintaining the differentiated state of the cell.
So, what could have been predicted from these facts?
That aging is caused by nothing more than cells losing their differentiation or becoming de- differentiated as I state in the title of this article. In reality, this should have been predicted long ago after studying Werner’s Syndrome and progeria. This prediction could have easily been made in 2014 and probably earlier after studies came out showing that Lamin A protein was involved in maintaining cellular differentiation in stem cells.
See> Gerontology. 2014;60(3):197-203. Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review
Take a skin cell for example, as it loses the factors that are suppressing genes that are not involved with being a skin cell, the cell starts adopting a more and more unusual (undifferentiated) phenotype.
If the process were to continue long enough the skin cell would be unrecognizable eventually. In some ways you could say the skin cell is returning to its undifferentiated, earlier (younger) state, but in an unhealthy way that ends up killing the bearer of these undifferentiated cells throughout the body. Counterintuitively, detrimental aging appears to actually be caused by cells becoming younger in a way, less differentitated, more like an embryonic stem cell!
I theorized in my 1998 paper, that more primitive organisms , early in evolution, probably reproduced in this manner- a quote-
“At this point in evolution, reproduction likely occurred through parthenogenesis and possibly the complete dissociation of the multi-celled organism into a myriad of single cell, clonal spores; in an unrestricted environment, this would provide a great reproductive advantage.”
And it turns out that there are still animals on earth that can reproduce this way..take the immortal jellyfish for example:
From National Geographic Magazine-
How the Jellyfish Becomes “Immortal”
“Turritopsis typically reproduces the old-fashioned way, by the meeting of free-floating sperm and eggs. And most of the time they die the old-fashioned way too. But when starvation, physical damage, or other crises arise, “instead of sure death, [Turritopsis] transforms all of its existing cells into a younger state,” said study author Maria Pia Miglietta, a researcher at Pennsylvania State University.The jellyfish turns itself into a bloblike cyst, which then develops into a polyp colony, essentially the first stage in jellyfish life.The jellyfish’s cells are often completely transformed in the process. Muscle cells can become nerve cells or even sperm or eggs.Through asexual reproduction, the resulting polyp colony can spawn hundreds of genetically identical jellyfish—near perfect copies of the original adult.”
It appears that our human development/ aging program of increasing differentiation then decreasing differentiation probably evolved from this ancient form of a reproduction system. Instead of a human dissolving into 30 trillion identical clonal spores to reproduce (which one would expect to happen if the selfish gene theory of evolution was the only way evolution worked), we instead lose our cellular differentiation in a way that harms and eventually kills. We might go so far to say that we age by getting younger from a differentiation point of view!-talk about an unexpected conclusion!
If this concept is correct then we can expect that with the addition of a number of healthy transcription factors back to an older cell that it could be made younger, and this has indeed been proven to be the case. All that was needed were the four transcription factors known as Yamanaka factors to reverse aging in the cell dramatically. The first experiment with Yamanaka factors took an adult cell and reprogrammed it all the way back to an embryonic state. They later just subjected an adult cell to transient expression of the Yamanaka factors and were able to make the cell significantly younger, but not return all the way to embryo status.
See> Cell. 2016 Dec 15; 167(7): 1719–1733.e12.In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming
“Aging is the major risk factor for many human diseases. In vitro studies have demonstrated that cellular reprogramming to pluripotency reverses cellular age, but alteration of the aging process through reprogramming has not been directly demonstrated in vivo. Here, we report that partial reprogramming by short-term cyclic expression of Oct4, Sox2, Klf4, and c-Myc (OSKM) ameliorates cellular and physiological hallmarks of aging and prolongs lifespan in a mouse model of premature aging. Similarly, expression of OSKM in vivo improves recovery from metabolic disease and muscle injury in older wild-type mice. The amelioration of age-associated phenotypes by epigenetic remodeling during cellular reprogramming highlights the role of epigenetic dysregulation as a driver of mammalian aging. Establishing in vivo platforms to modulate age-associated epigenetic marks may provide further insights into the biology of aging.”
See Update #23 with a link to a new Horvath study in press where he determined that this technique of transient expression of Yamanaka Factors that was applied to old rats reversed the aging in their cells by 54% to 75% depending on the tissue type, and had a major rejuvenating effect on them!
Okay, so here is another a little prediction that could be made:
If aging is caused by the loss of differentiation in your cells, then one would expect to see genes that produce transcription factors shut down. Also, thinking back to Werner’s Syndrome and progeria we would also expect to see some sort of pro-aging related protein unleashed that leads to truncated differentiation proteins like Lamin A and WRN.
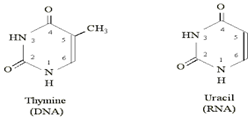
What kind of gene product would we be looking for that truncates differentiation proteins? The easiest way to truncate proteins would not be at the protein level, but rather at the mRNA level. The way proteins are produced is that the genes in our DNA are read and copied to a very similar molecule called mRNA which is almost identical to DNA with the exception of using the base pair Uracil in place of Thymine in the GCAT alphabet of your DNA. The only difference between Uracil and Thymine is a single methyl group (CH3) which is found attached to the 5’ carbon in thymine but only an H is attached to the 5’ carbon in uracil.
Prediction: There should exist some sort of protein that truncates mRNA transcripts at inappropriate places that increase with age to cause impairment of various differentiation proteins like WRN and Lamin A. This would be a lot easier that cutting the proteins after they have already been made. In fact, I did make this prediction to a pair of researchers who were able to rejuvenate old mice by removing half their blood plasma and replacing it with saline and albumin. I suggested they look for an aging-promoting RNA-ase that ran around truncating mRNA transcripts in inappropriate places-I never heard back from them.
Well as mentioned before, LARP1 seems to fit the description of this hypothetical protein! It has a very unique sequence that is specific for binding to RNA transcripts. It is found at high levels in cancers. Werner’s Syndrome victims suffer from normal cancers at a high rate as well- is LARP1 cleaving the WRN protein which leads to cancer?
So, the bottom-line conclusions we can draw from this amazing new study are these:
- Because the large set of genes shut down by methylation during the aging process (as well as the upregulated LARP1 gene( a true aging gene) ) are primarily the same across all mammalian species it very, very, strongly suggests that aging evolved and is highly conserved. This is in complete contradiction to modern mainstream evolutionary theory which proclaims that aging could never have evolved because it is bad for the individual and reduces the spread of the individual’s genes. For most modern aging theorists, they think an evolved aging program is something akin to a perpetual motion machine, completely impossible. In fact, this was once the quote by Aubrey De Grey in his sophomoric paper about how programmed aging was impossible.
See> Calorie restriction, post-reproductive life span, and programmed aging: a plea for rigor. Grey AD, Ann N Y Acad Sci. 2007 Nov;1119:296-305.
Please Notice I did not cross out the hormonal/neuroendocrine theory of aging…that one is still valid and will be found to be the upstream controller of the programmed loss of cellular differentiation primarily through the large/dramatic post age 50 increases in LH, FSH, and hCG with the simultaneous dramatic decline in night time melatonin peaks, dhea, pregnenolone, and progesterone.
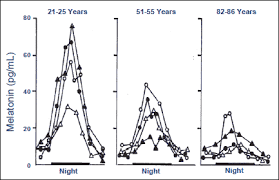
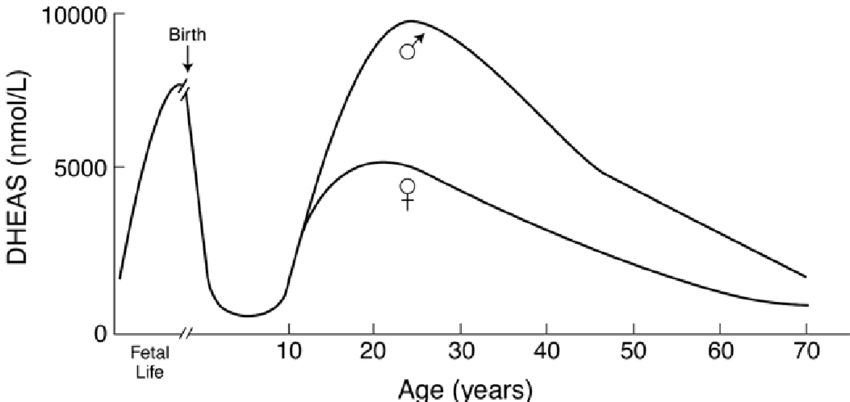
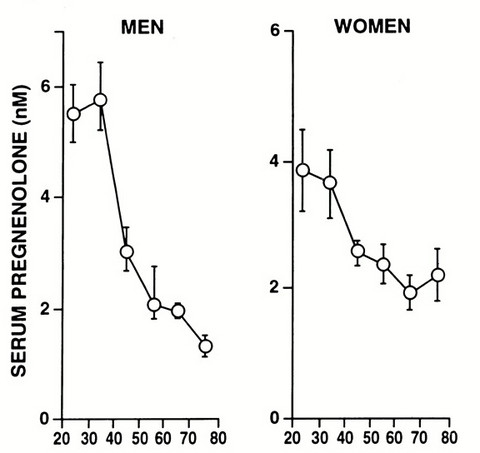
Interestingly, melatonin has been found to do all sorts of amazing things, like reversing recent onset menopause (probably due to melatonin’s ability to suppress LH and FSH), preventing the progression of Alzheimer’s, increasing dramatically during caloric restriction, acting as birth control in women at 75 mg per night, and even extending the lives of mice by 20%. I can easily imagine that melatonin somehow has a central role in maintaining the methylated status of the circadian rhythm and Alzheimer’s genes that become hypomethylated during aging (Horvath found these in the small group of genes that get activated with aging along with LARP1).
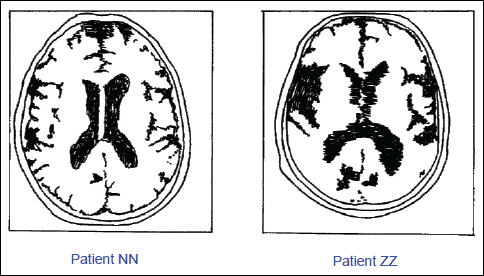
Fig. 8. Comparison of MNR brain recording of monozygotic twins, both of them were suffering from the Alzheimer's disease. The patient on the left (NN) was given melatonin (6 mg/day) for the period of 36 months, whereas patient on the right (ZZ) was given a placebo. Note the bitemporal atrophy and an enlargement of ventricules in the non-treated patient on the right (ZZ). (Adapted from Brusco et al. 1998).
A quick Pub Med search of the terms “melatonin AND DNA AND methylation” gives you 96 studies , most of which show that melatonin is intricately involved with DNA methylation, and the decline of melatonin with age might be the reason for the global hypomethylated status of DNA in the elderly. Studies with titles such as>> Melatonin and sirtuins: A “not-so unexpected” relationship., or Neuroendocrine aging precedes perimenopause and is regulated by DNA methylation Melatonin-induced demethylation of antioxidant genes increases antioxidant capacity through RORalpha in cumulus cells of prepubertal lambs, Melatonin-Mediated Development of Ovine Cumulus Cells, Perhaps by Regulation of DNA Methylation, Melatonin restores the pluripotency of long-term-cultured embryonic stem cells through melatonin receptor-dependent m6A RNA regulation (of Yamanaka factors) are not uncommon.
Likewise, a few studies have recently shown that DHEA also affects DNA methylation and DNA methyltransferase activity>> Epigenetic Age Reversal by Cell-Extrinsic and Cell-Intrinsic Means. A DNA Methylation Signature of Addiction in T Cells and Its Reversal With DHEA Intervention. Ethnic differences in DNA methyltransferases expression in patients with systemic lupus erythematosus.
There are numerous studies showing that progesterone, testosterone, and estrogen have dramatic effects on DNA methylation.
I also did not cross out the telomere theory of aging. However, this theory becomes just a subset of the loss of cellular differentiation theory of aging in that telomeres when they are long, fold back over on the coding DNA and suppress various genes, probably aging genes. As the telomere shortens, these genes are then expressed. This is called the telomere position effect. (Maybe the genes that , when expressed, lead to the methylation/suppression of those 36 transcription factor genes might be found here-( However I kind of doubt it because mice have been shown to have no increase in aging symptoms after their telomerase gene is knocked out and they have continually shortening telomeres. The only aging symptoms that show up in the experimental mice occur in the 4th generation when they start showing hair-graying, alopecia, and infertility-see DePinho)). (Update! Stay tuned..it turns out that TET enzymes are the likely suspects involved with the methylation of the 36 genes-to be discussed later).
Also, Horvath noted that some other aging genes that were hypomethylated and thus activated were a group of genes involved with the circadian rhythm. This suggests to me a connection to melatonin and other hormones that vary throughout the day. He noted another set of genes involved with causing Alzheimer’s disease that also lose methylation and are more highly expressed. (This might explain why melatonin seems so effective at stopping the progression of Alzheimer’s).
So of course this study raises the important question-What is the purpose of the evolution of programmed aging and how could it evolve?
A brief article about how evolution can select FOR aging>>
A Unifying Theory of The Evolution of Sex and Aging Via Predator Selection
Or a more in depth book on the topic>>>>
Update 1. The recent study where 50% of the blood plasma of mice was replaced with saline and albumin which led to a dramatic rejuvenation of the mice earlier was suggested herein to possibly be caused by a reduction of the LARP1 protein. However, what if LARP1 protein does not circulate in the blood but is only found inside the cell? What else could be being removed from the blood that stops the aging process and allows rejuvenation to happen? How about a 50% reduction in the circulating gonadotropins LH, FSH, and hCG ? These are the pro-aging hormones that increase with age by hundreds of percent and even up to 1,000% in women and men after age 50.
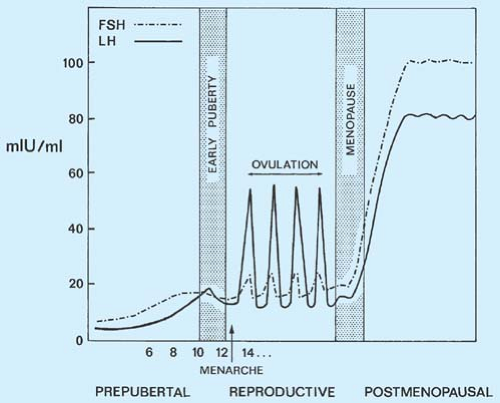
Update 2. It is interesting to note how babies often look so much alike due to their not being fully “differentiated”. They are much more unique and differentiated as children and adults. But then think of the elderly; don’t they seem to be very similar looking? Is this an example of a gain then a loss of cellular differentiation manifesting itself in physical appearance?

Update 3- It appears this aging system is kind of a case of antagonistic pleiotropy (AP). How? It is a new kind of AP where something that was good for your distant ancestors (dissolution of the organism into millions of billions of single cell clones that can each grow into a new adult) from an evolutionary perspective, evolves into something that is bad for the more modern descendants of the ancestral species. The ancient, dramatically prolific system of reproduction has evolved into something that now kills an individual at a programmed time.
Update 4– If progeria and Werner’s Syndrome are both manifestations of a malfunctioning development/differentiation program we can make two very important observations:
- The genes silenced by the Lamin a protein that is defective in progeria, are most likely the same genes that control the developmental changes where an infant develops into a prepubescent juvenile. (Keep in mind that progeria begins at birth) and
- The genes silenced by the WRN protein that is defective in Werner’s Syndrome, are most likely the genes that control the developmental changes where a prepubescent juvenile develops into a fully sexually developed/differentiated fertile adult. (Remember that Werner’s Syndrome does not kick in until puberty begins).
Update 5- For decades if not a century, evolutionary biologists and gerontologists, in order to maintain the illusion that aging is not programmed and was not selected for have had to hide certain facts that just screamed out “aging is programmed!”. The two diseases that are the main topic of this article, Werner’s Syndrome, and progeria have been referred to over and over as “not real examples of aging” because they are caused by genetic mutations and have a few differences when compared to regular aging . This is especially true in the case of Werner’s disease where a 50 year old woman will look almost identical to a normally aging 85 or 90 year old woman! Because Werner’s Syndrome patients have a higher incidence of some rare cancers – scientists of the past have relied on this canard to declare Werner’s Syndrome is not a case of accelerated programmed aging. With the programmed loss of cellular differentiation theory of aging we can finally do away with this pretense.
Another glaring fact that screams “aging is programmed” is the existence of semelparous aging. The most famous example consists of the rapid aging and death of the Pacific Salmon immediately after breeding around the age of three years old. When the Pacific Salmon is castrated , it can live 7 or more years. How was this explained using the other theories of aging of the past? Simply by saying semelparous aging is not a real form of aging and can be ignored! It had long been hypothesized that the rapid aging and death of the Pacific Salmon was caused by its huge exertion of energy and large amounts of resources spent in traveling the long journey from the ocean to its riparian birthplace to reproduce. Some suggested that the Salmon just died of exhaustion. Others made a slightly less simple case and suggested that the hormonal changes occurring during the great trip of the Salmon led to very high levels of the stress hormone cortisol. Supposedly that was what was killing them, although the cortisol increase comes long before they reproduce.
Well, recently Craig Atwood did a study of changes in the pro-aging hormones LH and FSH various species including the semelparous Salmon. Although apparently he could not find any data on changes in LH levels in Salmon he did find that FSH levels skyrocket 4,500% post reproduction as compared to even the high level reached in humans after age 50 of about a 500% increase.
I would like to point out to Craig that there is a 1998 study on hormone changes in Salmon that shows LH levels skyrocket as well. Biol Reproduction 1998 Mar;58(3):814-20. B. Borg et. al.
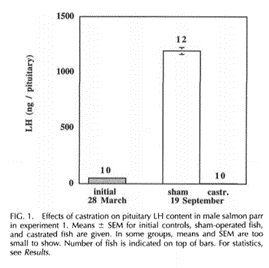
So the bottom line here is we see that semelparous species no longer have to be put in a special category and hidden away and ignored as not related to normal aging. Rather they now provide a somewhat typical case of programmed aging being driven by the post reproductive dramatic increases in FSH and LH seen also in humans and most other animals from fish to mammals to birds, etc. The salmon are only unusual in the speed at which they age and the height to which their LH and FSH levels can reach.
Update 6-
Now this one really gets into the weeds of this theory-probably not suitable for the casual reader:
It is interesting to consider these two abstracts concerning menopause, hormones and methylation>
Significance
Within an evolutionary framework, aging and reproduction are intrinsically linked. Although both laboratory and epidemiological studies have observed associations between the timing of reproductive senescence and longevity, it is not yet known whether differences in the age of menopause are reflected in biomarkers of aging. Using our recently developed biomarker of aging, the “epigenetic clock,” we examined whether age at menopause is associated with epigenetic age of blood, saliva, and buccal epithelium. This is a definitive study that shows an association between age of menopause and biological aging (measured using the epigenetic clock). Our results also indicate menopause may accelerate the epigenetic aging process in blood and that age at menopause and epigenetic age acceleration share a common genetic signature.
2019 Feb;74:213-224.
Neuroendocrine aging precedes perimenopause and is regulated by DNA methylation
Abstract
Perimenopause marks initiation of female reproductive senescence. Age of onset is only 47% heritable suggesting that additional factors other than inheritance regulate this endocrine aging transition. To elucidate these factors, we characterized transcriptional and epigenomic changes across endocrine aging using a rat model that recapitulates characteristics of the human perimenopause. RNA-seq analysis revealed that hypothalamic aging precedes onset of perimenopause. In the hypothalamus, global DNA methylation declined with both age and reproductive senescence. Genome-wide epigentic analysis revealed changes in DNA methylation in genes required for hormone signaling, glutamate signaling, and melatonin and circadian pathways. Specific epigenetic changes in these signaling pathways provide insight into the origin of perimenopause-associated neurological symptoms such as insomnia. Treatment with 5-aza-2′-deoxycytidine, a DNA-methyltransferase-1 inhibitor, accelerated transition to reproductive senescence/ whereas supplementation with methionine, a S-adenosylmethionine precursor, delayed onset of perimenopause and endocrine aging. Collectively, these data provide evidence for a critical period of female neuroendocrine aging in brain that precedes ovarian failure and that DNA methylation regulates the transition duration of perimenopause to menopause.
Update 8-
Once evolutionary biologists realize that there is something more going on to drive evolution than the selfish gene, once they realize there are evolutionary forces that also limit the spread of an individual’s genes for the good of the species which I describe in my article “Sex & Aging , How Evolution Selects For Them Almost Everywhere All the Time” many enduring mysteries of evolution can be easily explained. For example, here is a large portion of the chapter on homosexuality in my book “What Darwin Could Not See- The Missing Half of the Theory”:
CHAPTER 6: The Sixth Puzzle Piece-Homosexuality In Animals & Humans
One would think that anyone defending the primacy of the selfish gene as the major driving force behind all of evolution would have some sort of reasonable explanation for how something as widespread as human homosexuality could evolve.
Homosexuality is a condition where the possessor of the homosexual trait will, if left to nature only, will choose to never have sex with the opposite sex and thus not have any offspring and not pass on a single gene to the gene pool! Yet human homosexuality exists and has persisted throughout history.
Certainly, this must be harder for selfish gene promoters to explain than sex. At least with sex, the reproducer gets to pass on half of his or her genes. Here the selfish gene-ist has to explain how it evolved that someone passes on NO GENES WHATSOEVER! Given the very tough problem to solve here, the topic of homosexuality is just ignored for the most part, by evolutionary biologists.
Richard Dawkins, as courageous as he is, at least gives an explanation a try in a 2015 YouTube video. I share the link with you below. If you want a good laugh give it a watch. I am not laughing at Dawkins himself just at him trying to perform the impossible task of explaining homosexuality from the selfish gene point of view.
Darwin Day 2015 Questions: #4 How does evolution explain homosexuality?
Richard Dawkins Foundation for Reason & Science

After viewing this I think Dawkins might seem to focus more on male than female homosexuality and was so bold as to suggest that bottle feeding babies (male I presume) might make them more inclined to be homosexual. I am guessing he thinks sucking on a rubber nipple trains the young lad to want to suck on other protuberances?
Biologists in general tend to also discuss the evolutionary puzzle of homosexuality as mainly a human condition. Applying it to just humans makes the fact conveniently unique and a special category that can be ignored. They do it all the time. Menopause and suicide are also promoted by most biologists as being exceptions that apply to just humans.
Well it turns out that humans aren’t the only ones where homosexuality is common.
There are some estimates that up to 1500 species have been documented to have homosexual individuals in their numbers! If you just do a quick perusal of the Wikipedia entry for homosexuality in animals, you will get all sorts of examples.
So apparently, homosexuality cannot be dumped into the unique human exception category and thus can no longer be ignored by biologists. It must be addressed; trying to address it from the perspective of the preeminence of the selfish gene just leads us into another blind alley with no way out.
What the Wikipedia entry fails to describe are the conditions affecting the pregnant mother of future homosexual offspring.
A number of studies in rodents have shown that if you stress the pregnant mother at certain times during her pregnancy she will tend to give birth to homosexual males and promiscuous females and a smaller number of homosexual females.
What is this telling us from the perspective of the BIG PICTURE? What causes stress? Too many close encounters with predators. This fits quite easily into the BIG PICURE of most unexplained biological phenomenon as being defenses to evolving predation.
How is having homosexual offspring a defense to predation? Having homosexual offspring is a form of birth control for mothers who are considered by evolution to be unfit in the presence of predation. The stress from predator encounters if extreme enough can kill the mothers and their unborn babies. If the stress is less extreme it can lead to homosexual offspring that need to be nursed for a relatively significant period of time. Nursing prevents the mother from becoming fertile for mating. So, in a sense having homosexual offspring is just nature’s form of birth control for mothers perceived as temporarily “unfit” due to stress.
Let us consider the case of female offspring of stressed mothers being more promiscuous than the female offspring of non-stressed mothers. This also jibes well with the BIG PICTURE as promiscuous females who have offspring from multiple males rather than bonding with a single one will add more diversity to the gene pool than if she just mated with a single male for life. As we will see later diversity in the gene pool is the defense to evolving predation that evolution seeks with all these mysterious adaptations.
Now we get to humans; is there any evidence that stressing pregnant female humans can cause their male offspring to be born as homosexual? I wrote about this topic in my December 2000 paper published in Medical Hypotheses titled “Sex, Kings, & Serial Killers and other Group Selected Traits”
Here is the excerpt:
Homosexuality: Birth Control for “Unfit” Mothers?
Prevailing evolutionary theory cannot explain the conundrum of homosexuality. Current theory requires defining homosexuality as an evolutionary accident as homosexual offspring would not be expected to reproduce. Is evolution so sloppy that the sexual preferences of 10% to 20% of the human population (78) is simply a random mistake of nature? And why does it also occur throughout the animal kingdom from sheep (79) on down to rats (80)? If one accepts group selection as a reality, the purpose of homosexuality has a simple explanation.
Various studies show that when stressed at a certain time during gestation, rats give birth to males that exhibit female behavior and females that are more masculine (81). (The literature is relatively conclusive on this for males, but the data on females is somewhat ambiguous. Some female offspring of stressed rats also show more promiscuous mating behavior). Stress increases cortisol levels in rats, and the Prior Paper referred to studies showing that cortisol appears to oppositely affect the sex hormones in human females and males which we will assume extends to rats.
If stress induces high maternal cortisol levels during gestation and the cortisol reaches the developing embryo, endogenous embryonic sex hormones may be altered. Testosterone and estradiol levels in male and female embryos respectively may be decreased. Decreased embryonic sex hormones likely affect the development of the brain’s sexuality. It has been shown that the prenatal stress-induced feminization of male rats is prevented by perinatal androgen treatment (82).
Studies have shown that human females, male transsexuals, and homosexuals share similarities in certain brain structures which differ with heterosexual males (83, 84). Also, it is believed that testosterone derived DHT is required during fetal brain development to create a “male brain” (85). Likewise, we might assume that estradiol exposure creates a “female brain” by feminizing some brain structures. If a stress-induced maternal cortisol surge suppresses the embryo’s testosterone or estradiol, then homosexual offspring, of either sex could result. Interestingly, some researchers found that in a large group of homosexuals interviewed in Germany, many more were born during the war years of 1941 to 1947 than before or after this stressful period with the birth peak occurring in 1944-1945 (86).
Why would evolution create such a system? If a pregnant female is stressed in the wild, it may imply close encounters with predators or maladaptation to her group. Evolution, through group selection, has likely selected for groups that remove or inhibit the spread of her “less fit” genes. While a spontaneous miscarriage or stressed-induced cannibalization of her young (which is common in rodents) is a simple solution, it would leave the female ready to reproduce again. A more clever and effective solution is to give her effectively sterile offspring which she will raise, and which will keep her from reproducing much longer than if she were childless. Also, if group survival required the homosexual children to reproduce, homosexual females could be forced to have sex by dominant heterosexual males. Homosexual males, however, who could not be forced, are evolutionarily irrelevant anyway as long as a single heterosexual male existed.
The only attempt at an evolutionary explanation of homosexuality that the author could find was one that proposed that a homosexual male child would be generated if it was prenatally stressed. The stressor was assumed to be the mother’s living in a crowded environment. The homosexual male, as an adult would not reproduce so that in times of famine there would be fewer grandchildren, and thus an increased likelihood of the grandchildren’s survival (87). One does not have to work long to find counter arguments to this reasoning, but it is a creative attempt to overcome the conundrum of homosexuality and borders on using group selection as an argument. It is only referenced here to show the difficulties that exist in trying to explain homosexuality without the unabashed acceptance of some form of group selection.
(Recent note-which we will later find this to be not group but an even higher-level form of selection called species selection which I have refined and coined the name “Predator Selection”).
One must wonder about the seemingly high levels of human homosexuality. Were so many mothers severely stressed by predators or wars during pregnancy? Not likely. However, a source of artificial stress has been unleashed this century on humans in epidemic proportions: cigarette smoking. Nicotine from smoking induces a significant increase in cortisol levels. If a pregnant female has the genetic predisposition to bear homosexual children when stressed, and she smokes during early pregnancy, the nicotine-induced cortisol increase may be sufficient to induce homosexuality in her offspring. This speculation could easily be confirmed or refuted with a simple epidemiological study.

- Sell R. Wells J. Wypij D. The prevalence of homosexual behavior and attraction in the United States, the United Kingdom, and France: results of national population-based samples. Archives of Sexual Behavior 24(3). 1995. 235-248.
- Perkins A. Fitzgerald J. Moss G. A comparison of LH secretion and brain estradiol receptors in heterosexual and homosexual rams and female sheep. Hormones & Behavior 29(1). 1995. 31-41.
- Ferguson T. Alternative sexualities in evolution. Evolutionary Theory 11(1). 1995. 55-64
- Ohkawa T. Sexual differentiation of social play and copulatory behavior in prenatally stressed male and female offspring of the rat: the influence of simultaneous treatment by tyrosine during exposure to prenatal stress. Nippon Naibunpi Gakkai Zasshi-Folia Endocrinolgica Japonica. 63(7):823-35, 1987 Jul.
- Dorner G. Gotz F. Docke W. Prevention of demasculization and feminization of the brain in prenatally stressed male rats by perinatal androgen treatment. Experimental & Clinical Endocrinology. 81(1):88-90 1983 Jan.
- Swaab D. Gooren L. Hofman M. Gender and sexual orientation in relation to hypothalamic structures. Hormone Research. 38 Suppl 2:51-61, 1992.
- Zhou J, Hofman M. Gooren L. Swaab D. A sex difference in the human brain and its relation to transsexuality. Nature. 378(6552):68-70, 1995 Nov.
- Connolly P. Choate J. Resko J. Effects of endogenous androgen on brain androgen receptors of the fetal rhesus monkey. Neuroendocrinology. 59(3):27 1994 Mar.
- Dorner G. et.al. Prenatal stress as possible aetiogenetic factor of homosexuality in human males. Endokrinologie.75(3):365-8, 1980 Jun.
- 81.
-END Excerpt from my 1998 paper-
(A natural born homosexual?)
While searching for the old studies that showed a sharp rise in the birth of homosexuals in Germany during the WWII years (this supposedly also happened in England as well amongst the pregnant women who hid in London’s subway tunnels during the German bombing campaigns) I found an article about a new book by Dr. Dick Swaab, a well-known neuroscientist who is best known for his research and discoveries in the field of brain anatomy and physiology, in particular the impact that various hormonal and biochemical factors in the womb have on brain development. The book is called “We Are Our Brains” and he puts forth the controversial “new” idea that homosexuality occurs in the brains of fetuses in the womb of stressed mothers. He also notes, like I did in my 2000 paper, that smoking by pregnant mothers can lead to homosexual offspring because nicotine stimulates the release of the stress hormone cortisol and in effect acts as a predator encounter as perceived by evolution. He does add some new evidence that stress in mothers causes homosexuality by noting, as one would expect, that amphetamines (also fake stress) and various other substances lead to an excess of homosexual offspring.
So, that’s about it for homosexuality. If we have learned another fact about evolution, we can say that homosexuality fits in to the big picture as follows:
-Homosexuality is birth control for mothers perceived as stressed by evolution and thus possibly having less than optimal genes for the particular environment. Having an effectively “sterile” child reduces her potential contribution to the gene pool by preventing her from getting pregnant while nursing, and investing resources into the sterile child which also reduces her total potential reproductive output.
One more Thing-Dr. Dick Swaab, for some reason is getting death threats from some gay people who don’t like homosexuality being portrayed as a pathology! I guess they want to think it is a choice. But if you ask most gay people they are happy to say they were born that way. So please don’t send me any death threats thank you.
One other thing I have seemed to notice is that when you look at large groups of either homosexual males, or homosexual females, the males still tend to maintain their evolved desire to stand out and draw attention to themselves while groups of lesbians seem to act more like the camouflaged females of other species, who desire and have evolved to avoid attention. You can also do a crude test of this idea by searching google-images for group of gay men and then for group of lesbians and you will see the difference. Most of the pictures are along the lines that follow: (see pictures in the book).
Update 9- This is a good one!
While doing research for my 1998 paper- The Evolution of Aging a New Approach to an Old Problem of Biology
I studied all accelerated aging diseases of progeria, the segmental aging diseases caused by various mitochondrial defects, ataxia telangiectasia (AT), Cockayne syndrome (CS) , xeroderma pigmentosum (XP), and Werner’s syndrome and compared them with respect to what aging symptoms they had at an accelerated rate…and made a table to compare them! (see table bellow). You could call this the periodic table of accelerated aging symptoms.
I had designated Werner’s syndrome as the dominant aging system that coopted and controlled all the other aging systems-you will see why when you study the table- but basically because WS shows all the symptoms of normal human aging while other rapid aging diseases just show a segment..
It turns out, I believe, given the connection between development and aging we now understand thanks to Horvath, that the normal Werner’s syndrome protein apparently is also the coordinator and master regulator of all other development genes and transcription factors. The WRN protein does not do all the work itself, it has apparently coopted downstream transcription/differentiation factors and tells them when to be turned on and turned off. WRN is the master regulator of development.
Somehow WRN controls how and when Lamin A protein shuts down various genes in its purview, as well as the normal proteins that are defective in the mitochondrial diseases , AT, CS, and XP. WRN tells them all what to do and when.
It also now reveals , what I believe , is the master plan of how development is regulated and orchestrated as you will soon see- I had an inkling about it when I made the table but I never articulated it. Well hold onto your hats…here it comes>>
|
Aging System #4 Senescent Gene Expression: FSH/DHT driven, seen in men at higher rate. (co-opts #3) (and #1?)
|
Aging System #5A Somatic atrophy: Mitochondrial Apoptosis, LH/hCG driven, seen in women at a higher rate
(co-opts #2)
|
Aging System #5B Somatic atrophy: nDNA Fragmentation Apoptosis, LH/hCG driven, seen in women at a higher rate
|
Aging System # 6 Sex tissue atrophy:
estrogen/DHT driven, seen in women at higher rate (co-opts #4, #5, (and #1))
|
|
Progeria only. Defective Lamin A protein-truncated
|
Mitochondrial Myopathy (MM), NARP (N), CPEO (CP), MELAS (ME), MERRF (MR) , KSS (K), Dystonia (D), Leigh’s Syndrome (LS)
|
Ataxia Telangiectasia (AT), Xeroderma Pigmentosum (XP), Cockayne Syndrome (CS). Various Defective proteins.
|
Werner’s Syndrome. (WS),
Bloom’s Syndrome (BS), Defective DNA helicase protein WRN- truncated
|
|
Coxa Valga & necrosis of head of femur
|
|||
|
Dysplastic osteoporosis (growing bones)
|
|||
|
Symptoms of #4 co-opted by #6
|
Symptoms of #6 co-opted from #4
|
||
|
Atherosclerosis
|
Atherosclerosis-WS
|
||
|
Hypertension
|
Hypertension-WS
|
||
|
Gray Hair
|
Gray Hair-WS
|
||
|
Alopecia
|
Alopecia-WS
|
||
|
Calcification of Heart Valves
|
Calc. of Heart Valves-WS
|
||
|
Laryngeal Atrophy
|
Laryngeal Atrophy-WS
|
||
|
Loss of subcutaneous tissue
|
Loss of subcut. tissue-WS
|
||
|
Hypermelanosis of Skin
|
Hypermelanosis of Skin-WS
|
||
|
Hypogonadism (defect of development?)
|
Hypogonadism -AT, XP
|
Hypogonadism -WS, BS
|
|
|
Symptoms of #5A also seen in #5B and co-opted by #6
|
Symptoms of #5B also seen in #5A and co-opted by #6
|
Symptoms of #6 co-opted from #5A and #5B
|
|
|
Muscle Wasting-MM, N
|
Muscle Wasting-AT
|
Muscle Wasting-WS
|
|
|
Neuronal Degeneration/Brain Atrophy-CP, ME, MR, K
|
Neuronal Degeneration/Brain Atrophy -AT, XP
|
Neuronal Degeneration, Brain Atrophy -WS
|
|
|
Basal Ganglion Calcification – D, LS
|
Basal Ganglion Calcification – CS
|
Basal Ganglion Calcification -WS
|
|
|
Cataracts-K
|
Cataracts-CS
|
Cataracts-WS
|
|
|
Diabetes-K
|
Diabetes-AT
|
Diabetes-BS, WS
|
|
|
Alzheimer’s Disease-mitochondrial induced
|
Alzheimer’s Disease-XP
|
Alzheimer’s Disease-WS
|
|
|
Symptoms of #5B co-opted by #6
|
Symptoms of #6 co-opted from #5B
|
||
|
Poor Healing -XP
|
Poor Healing -WS
|
||
|
Skin Ulcers -XP
|
Skin Ulcers -WS
|
||
|
Thymic Atrophy-AT
|
Thymic Atrophy-BS, WS
|
||
|
Scaly Skin-XP
|
Scaly Skin-WS
|
||
|
Somatic Cancers-XP,AT
|
Somatic Cancers- BS, WS
|
||
|
Lipofuscin Accumulation-CS,XP
|
Lipofuscin Accumulation-WS
|
||
|
Arthritis-AT
|
Arthritis-WS
|
||
|
Peripheral Osteoporosis-CS
|
Peripheral Osteoporosis-WS (growth plate closure) maybe unique to #6?
|
||
|
Symptoms unique to #6
|
|||
|
Menopause-WS
|
|||
|
Breast, Uterine, and Ovarian atrophy and cancer-WS
|
|||
|
Prostate atrophy-WS, hyperplasia-WS, and cancer-WS
|
|||
|
Depression-WS?
|
An interesting coincidence is that from this table we see there appears to be 4 unique development/aging programs for 4 different groups of tissue types. Does this somehow relate to the fact that there are 4 Yamanaka factors that can reverse aging? I will be looking into this.
So that’s it for update #9!
Update #10:
One thing I am wondering about…
The discovery of Yamanaka factors that can age somatic cells in reverse should not have come as much of a surprise. Why? They should have been predicted the day it was announced that Dolly the sheep was cloned. In her case an adult somatic nucleus was inserted into an ovum, zapped with a little electricity and the nucleus reprogramed itself all the way back to a single cell undifferentiated embryo. What is being done with Yamanka factors is the same thing that has been going on inside cloned embryos since the 1990’s. The real question here is what took so long to discover the Yamanaka factors?
Update #11
This update discussion has been deleted I am still working on it but I provide an interesting abstract to consider
J Gerontol A Biol Sci Med Sci
.2019 Aug 16;74(9):1391-1395.
Centenarians Overexpress Pluripotency-Related Genes
Marta Inglés 1, 2, José Viña 2
Abstract
Human mesenchymal cells can become pluripotent by the addition of Yamanaka factors OCT3/4, SOX2, c-MYC, KLF4. We have recently reported that centenarians overexpress BCL-xL, which has been shown to improve pluripotency; thus, we aimed to determine the expression of pluripotency-related genes in centenarians. We recruited 22 young, 32 octogenarian, and 47 centenarian individuals and determined the mRNA expression of Yamanaka factors and other stemness-related cell surface marker genes (VIM, BMP4, NCAM, BMPR2) in peripheral blood mononuclear cells by reverse transcription polymerase chain reaction. We found that centenarians overexpress OCT3/4, SOX2, c-MYC, VIM, BMP4, NCAM, and BMPR2, when compared with octogenarians (p < .05). We further tested the functional role of BCL-xL in centenarians’ ability to express pluripotency-related genes: lymphocytes from octogenarians transduced with BCL-xL overexpressed SOX2, c-MYC, and KLF4. We conclude that centenarians overexpress Yamanaka Factors and other stemness-related cell surface marker genes, which may contribute to their successful aging.
Update #12 Turns out lamin A is missing in undifferentiated embryonic stem cells and is defective in the rapid aging disease of progeria :
Efficient induction of pluripotent stem cells from granulosa cells by Oct4 and Sox2
Jian Mao 1, Qian Zhang, Xiaoying Ye, Kai Liu, Lin Liu
Abstract
Various types of somatic cells can be reprogrammed to induced pluripotent stem (iPS) cells. Somatic stem cells exhibit enhanced reprogramming efficiency by fewer factors, in contrast to fully differentiated cells. Nuclear Lamin A is highly expressed in differentiated cells, and stem cells are characterized by the absence of Lamin A. Granulosa cells (GCs) and cumulus cells in the ovarian follicles effectively and firstly generated cloned mice by somatic cell nuclear transfer, and these cells lack Lamin A expression. We tested the hypothesis that GCs could be effectively used to generate iPS cells with fewer factors. We show that iPS cells are generated from GCs at high efficiency even with only two factors, Oct4 and Sox2, like the iPS cells generated using four Yamanaka factors. These iPS cells show pluripotency in vitro and in vivo, as evidenced by high expression of pluripotency-associated genes, Oct4, Nanog, and SSEA-1, differentiation into three embryonic germ layers by embryoid body formation and teratoma tests, as well as high efficient generation of chimeras. Moreover, the exogenous genes are effectively silenced in these iPS cells. These data provide additional evidence in supporting the notion that reduced expression of LaminA and stem cells can improve the reprogramming efficiency to pluripotency.
Update #13
I wrote in my 1998 paper > ” Cancer, in a broad sense, may simply be a cell returning to its earlier, primitive, immortalized, state. It should not be very surprising that a mortal life form that evolved from a previously immortalized life form could spontaneously become immortalized through loss of some type of control. However, if the mortal life form had evolved from mortal ancestors, spontaneous immortalization would seem to be quite a miracle indeed.” see comment in full context at the end of this update .
Well, well, well>> I have been reading all the abstracts in Pub Med that contain the term “Yamanaka Factors” and what have I found? The cancer cells are basically just malfunctioning de-differentiated embryonic stem cells. And yes it seems very likely they are simply a reemergence of our oldest ancestors…Single cell life that lived before the age of oxygen. Both cancer cells and embryonic stem cells can divide indefinitely (immortal). Both of the them do not use oxygen for energy even when oxygen is present but rather switch to an anaerobic form of glycolysis for energy ! What follows are the interesting abstracts that show how this view of cancer being a reversion of cells to their most primitive state seems to be correct:
IT LOOKS LIKE WHAT WE ARE SEEING IN EMBRYONIC STEM CELLS AND CANCER CELLS IS THE PRIMORDIAL ANCESTOR OF ALL LIFE FORMS PREDICTED TO EXIST BY DARWIN-APPEARING AT GROUND 0 OF THE CELLULAR PATH TO HUMAN DEVELOPMENT.
The role of pluripotency factors to drive stemness in gastrointestinal cancer
Abstract
A better molecular understanding of gastrointestinal cancers arising either from the stomach, the pancreas, the intestine, or the liver has led to the identification of a variety of potential new molecular therapeutic targets. However, in most cases surgery remains the only curative option. The intratumoral cellular heterogeneity of cancer stem cells, bulk tumor cells, and stromal cells further limits straightforward targeting approaches. Accumulating evidence reveals an intimate link between embryonic development, stem cells, and cancer formation. In line, a growing number of oncofetal proteins are found to play common roles within these processes. Cancer stem cells share features with true stem cells by having the capacity to self-renew in a de-differentiated state, to generate heterogeneous types of differentiated progeny, and to give rise to the bulk tumor. Further, various studies identified genes in cancer stem cells, which were previously shown to regulate the pluripotency circuitry, particularly the so-called “Yamanaka-Factors” (OCT4, KLF4, SOX2, and c-MYC). However, the true stemness potential of cancer stem cells and the role and expression pattern of such pluripotency genes in various tumor cell types remain to be explored. Here, we summarize recent findings and discuss the potential mechanisms involved, and link them to clinical significance with a particular focus on gastrointestinal cancers.
The oncogene c-Jun impedes somatic cell reprogramming
Oncogenic transcription factors are known to mediate the conversion of somatic cells to tumour or induced pluripotent stem cells (iPSCs).
EMBO Rep
. 2014 Mar;15(3):244-53.
Dedifferentiation and reprogramming: origins of cancer stem cells
Abstract
Regenerative medicine aims to replace the lost or damaged cells in the human body through a new source of healthy transplanted cells or by endogenous repair. Although human embryonic stem cells were first thought to be the ideal source for cell therapy and tissue repair in humans, the discovery by Yamanaka and colleagues revolutionized the field. Almost any differentiated cell can be sent back in time to a pluripotency state by expressing the appropriate transcription factors. The process of somatic reprogramming using Yamanaka factors, many of which are oncogenes, offers a glimpse into how cancer stem cells may originate. In this review we discuss the similarities between tumor dedifferentiation and somatic cell reprogramming and how this may pose a risk to the application of this new technology in regenerative medicine.
J Cell Sci
. 2013 Aug 15;126(Pt 16):3638-48.
Abstract
Induced pluripotent stem cells (iPSCs) hold great promise for cell therapy. However, their low efficiency of lineage-specific differentiation and tumorigenesis severely hinder clinical translation. We hypothesized that reprogramming of somatic cells into lineage-specific progenitor cells might allow for large-scale expansion, avoiding the tumorigenesis inherent with iPSCs
Expert Rev Anticancer Ther
. 2021 Apr 8.
Pluripotency inducing Yamanaka factors: role in stemness and chemoresistance of liver cancer
Abstract
Introduction: Liver cancer is a major cause of mortality and is characterized by the transformation of cells into an uncontrolled mass of tumor cells with many genetic and epigenetic changes, which lead to the development of tumors. A small subpopulation of cell population known as Cancer Stem Cells (CSCs) is responsible for cancer stemness and chemoresistance. Yamanaka factors [octamer-binding transcription factor 4 (OCT4), SRY (sex-determining region Y)-box 2 (SOX2), kruppel like factor 4 (KLF4), and Myelocytomatosis (MYC); OSKM] are responsible for cancer cell stemness, chemoresistance, and recurrence.
Biochem Biophys Res Commun
. 2019 Sep 17;517(2):324-329.
Abstract
Induced pluripotent stem cells (iPSC) have a great potential, but their clinical application depends on finding strategies to abolish their tumorigenic potential. The use of Oct4, Sox2, Klf4, c-Myc and Nanog to generate iPSC demonstrated the already known importance of these genes to maintain stemness. Therefore, the presence of these genes is responsible for iPSC-derived teratomas. Similar to iPSC, P19 teratocarcinoma cell line also has characteristics of embryonic carcinoma cells and the ability to differentiate into many cell types. We separately silenced the transcription factors Oct4, Sox2, Klf4, c-Myc and Nanog in P19 cells and measured the impact of this silencing in vivo. All silenced cells generated tumors when injected in immunosuppressed mice, but silencing of Oct4, Sox2 and Klf4 generated mainly teratomas with mesoderm tissue. Our results suggest that downregulation of these transcription factors is not enough to avoid the formation of teratomas, but their silencing affect their differentiation potential.
Oncogene
. 2019 Aug;38(34):6226-6239.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) arises through accumulation of multiple genetic alterations. However, cancer cells also acquire and depend on cancer-specific epigenetic changes. To conclusively demonstrate the crucial relevance of the epigenetic programme for the tumourigenicity of the cancer cells, we used cellular reprogramming technology to reverse these epigenetic changes. We reprogrammed human PDAC cultures using three different techniques – (1) lentivirally via induction of Yamanaka Factors (OSKM), (2) the pluripotency-associated gene OCT4 and the microRNA mir-302, or (3) using episomal vectors as a safer alternative without genomic integration. We found that induction with episomal vectors was the most efficient method to reprogram primary human PDAC cultures as well as primary human fibroblasts that served as positive controls. Successful reprogramming was evidenced by immunostaining, alkaline phosphatase staining, and real-time PCR. Intriguingly, reprogramming of primary human PDAC cultures drastically reduced their in vivo tumourigenicity, which appeared to be driven by the cells’ enhanced differentiation and loss of stemness upon transplantation. Our study demonstrates that reprogrammed primary PDAC cultures are functionally distinct from parental PDAC cells resulting in drastically reduced tumourigenicity in vitro and in vivo. Thus, epigenetic alterations account at least in part for the tumourigenicity and aggressiveness of pancreatic cancer, supporting the notion that epigenetic modulators could be a suitable approach to improve the dismal outcome of patients with pancreatic cancer.
Methods Mol Biol
. 2019;1916:249-261.
Reprogramming of Human Melanocytes and Melanoma Cells with Yamanaka Factors
Abstract
The expression of Yamanaka factors (Oct3/4, Klf-4, Sox-2, c-Myc) can reprogram cancer cells to a pluripotent stage. This may cause the removal of their epigenetic memory and result in altered tumorigenicity. Various studies in the literature have shown that cancer cell reprogramming is a potential tool to study disease progression or discover novel therapeutic or diagnostic markers in cancer research. In this chapter, we aim to introduce the cancer cell reprogramming protocol in detail by using human melanocytes and melanoma cell lines, and Sendai viral vectors encoding Yamanaka factors have been used to reprogram cells. Representative results are discussed and important notes have been summarized in order to point out important steps during cancer cell reprogramming.
J Biomed Sci
. 2018 Jul 19;25(1):57.
Abstract
Background: Induced pluripotency in cancer cells by ectopic expression of pluripotency-regulating factors may be used for disease modeling of cancers. MicroRNAs (miRNAs) are negative regulators of gene expression that play important role in reprogramming somatic cells. However, studies on the miRNA expression profile and the expression patterns of the mesenchymal-epithelial transition (MET)/epithelial-mesenchymal transition (EMT) genes in induced pluripotent cancer (iPC) cells are lacking.
Methods: iPC clones were generated from two colorectal cancer (CRC) cell lines by retroviral transduction of the Yamanaka factors. The iPC clones obtained were characterized by morphology, expression of pluripotency markers and the ability to undergo in vitro tri-lineage differentiation. Genome-wide miRNA profiles of the iPC cells were obtained by microarray analysis and bioinformatics interrogation. Gene expression was done by real-time RT-PCR and immuno-staining; MET/EMT protein levels were determined by western blot analysis.
Results: The CRC-iPC cells showed embryonic stem cell-like features and tri-lineage differentiation abilities. The spontaneously-differentiated post-iPC cells obtained were highly similar to the parental CRC cells. However, down-regulated pluripotency gene expression and failure to form teratoma indicated that the CRC-iPC cells had only attained partial pluripotency. The CRC-iPC cells shared similarities in the genome-wide miRNA expression profiles of both cancer and pluripotent embryonic stem cells. One hundred and two differentially-expressed miRNAs were identified in the CRC-iPC cells, which were predicted by bioinformatics analysis be closely involved in regulating cellular pluripotency and the expression of the MET/EMT genes, possibly via the phosphatidylinositol-3 kinases-protein kinase B (PI3K-Akt) and transforming growth factor beta (TGF-β) signaling pathways. Irregular and inconsistent expression patterns of the EMT vimentin and Snai1 and MET E-cadherin and occludin proteins were observed in the four CRC-iPC clones analyzed, which suggested an epithelial/mesenchymal hybrid phenotype in the partially reprogrammed CRC cells. MET/EMT gene expression was also generally reversed on re-differentiation, also suggesting epigenetic regulation.
Conclusions: Our data support the elite model for cancer cell-reprogramming in which only a selected subset of cancer may be fully reprogrammed; partial cancer cell reprogramming may also elicit an epithelial-mesenchymal mixed phenotype, and highlight opportunities and challenges in cancer cell-reprogramming.
Biochim Biophys Acta Rev Cancer
. 2018 Jan;1869(1):1-10.
Deubiquitylating enzymes as cancer stem cell therapeutics
Abstract
The focus of basic and applied research on core stem cell transcription factors has paved the way to initial delineation of their characteristics, their regulatory mechanisms, and the applicability of their regulatory proteins for protein-induced pluripotent stem cells (protein-IPSC) generation and in further clinical settings. Striking parallels have been observed between cancer stem cells (CSCs) and stem cells. For the maintenance of stem cells and CSC pluripotency and differentiation, post translational modifications (i.e., ubiquitylation and deubiquitylation) are tightly regulated, as these modifications result in a variety of stem cell fates. The identification of deubiquitylating enzymes (DUBs) involved in the regulation of core stem cell transcription factors and CSC-related proteins might contribute to providing novel insights into the implications of DUB regulatory mechanisms for governing cellular reprogramming and carcinogenesis. Moreover, we propose the novel possibility of applying DUBs coupled with core transcription factors to improve protein-iPSC generation efficiency. Additionally, this review article further illustrates the potential of applying DUB inhibitors as a novel therapeutic intervention for targeting CSCs. Thus, defining DUBs as core pharmacological targets implies that future endeavors to develop their inhibitors may revolutionize our ability to regulate stem cell maintenance and differentiation, somatic cell reprogramming, and cancer stem cells.
Biochim Biophys Acta Mol Cell Res
. 2017 Jul;1864(7):1359-1369.
Abstract
Reprogramming, or generation of induced pluripotent stem (iPS) cells (functionally similar to embryonic stem cells or ES cells) by the use of transcription factors (typically: Oct3/4, Sox2, c-Myc, Klf4) called “Yamanaka factors” (OSKM), has revolutionized regenerative medicine. However, factors used to induce stemness are also overexpressed in cancer. Both, ES cells and iPS cells cause teratoma formation when injected to tissues. This raises a safety concern for therapies based on iPS derivates. Transdifferentiation (lineage reprogramming, or -conversion), is a process in which one mature, specialized cell type changes into another without entering a pluripotent state. This process involves an ectopic expression of transcription factors and/or other stimuli. Unlike in the case of reprogramming, tissues obtained by this method do not carry the risk of subsequent teratomagenesis.
Iran J Basic Med Sci
. 2016 Oct;19(10):1131-1135.
Abstract
Objectives: Similar characteristics of molecular pathways between cellular reprogramming events and tumorigenesis have been accentuated in recent years. Reprogramming-related transcription factors, also known as Yamanaka factors (OCT4, SOX2, KLF4, and c-MYC), are also well-known oncogenes promoting cancer initiation, progression, and cellular transformation into cancer stem cells. Long non-coding RNAs (lncRNAs) are a major class of RNA molecules with emerging roles in stem cell pluripotency, cellular reprogramming, cellular transformation, and tumorigenesis. The long intergenic non-coding RNA ROR (lincRNA-ROR, linc-ROR) acts as a regulator of cellular reprograming through sponging miR-145 that normally negatively regulates the expression of the stemness factors NANOG, OCT4, and SOX2.
Stem Cells
. 2016 Nov;34(11):2613-2624.
Positive Feedback Loop of OCT4 and c-JUN Expedites Cancer Stemness in Liver Cancer
Abstract
The network of stemness genes and oncogenes in human patient-specific reprogrammed cancer stem cells (CSCs) remains elusive, especially in liver cancer. HepG2-derived induced pluripotent stem cell-like cells (HepG2-iPS-like cells) were generated by introducing Yamanaka factors and the knockdown vector shTP53. They exhibited features of stemness and a higher tumorigenesis after xenograft transplantation compared with HepG2 cells. The cancerous mass of severe combined immunodeficiency (SCID) mice derived from one colony was dissected and cultured to establish reprogrammed HepG2-derived CSC-like cells (designated rG2-DC-1C). A single colony exhibited 42% occurrence of tumors with higher proliferation capacities. rG2-DC-1C showed continuous expression of the OCT4 stemness gene and of representative tumor markers, potentiated chemoresistance characteristics, and invasion activities. The sphere-colony formation ability and the invasion activity of rG2-DC-1C were also higher than those of HepG2 cells. Moreover, the expression of the OCT4 gene and the c-JUN oncogene, but not of c-MYC, was significantly elevated in rG2-DC-1C, whereas no c-JUN expression was observed in HepG2 cells. The positive-feedback regulation via OCT4-mediated transactivation of the c-JUN promoter and the c-JUN-mediated transactivation of the OCT4 promoter were crucial for promoting cancer development and maintaining cancer stemness in rG2-DC-1C. Increased expression of OCT4 and c-JUN was detected in the early stage of human liver cancer. Therefore, the positive feedback regulation of OCT4 and c-JUN, resulting in the continuous expression of oncogenes such as c-JUN, seems to play a critical role in the determination of the cell fate decision from iPS cells to CSCs in liver cancer. Stem Cells 2016;34:2613-2624.
Stem Cell Reports
. 2016 Jul 12;7(1):1-10.
MiR-31/SDHA Axis Regulates Reprogramming Efficiency through Mitochondrial Metabolism
Abstract
Metabolism is remodeled when somatic cells are reprogrammed into induced pluripotent stem cells (iPSCs), but the majority of iPSCs are not fully reprogrammed. In a shift essential for reprogramming, iPSCs use less mitochondrial respiration but increased anaerobic glycolysis for bioenergetics. We found that microRNA 31 (miR-31) suppressed succinate dehydrogenase complex subunit A (SDHA) expression, vital for mitochondrial electron transport chain (ETC) complex II. MiR-31 overexpression in partially reprogrammed iPSCs lowered SDHA expression levels and oxygen consumption rates to that of fully reprogrammed iPSCs, but did not increase the proportion of fully reprogrammed TRA1-60(+) cells in colonies unless miR-31 was co-transduced with Yamanaka factors, which resulted in a 2.7-fold increase in full reprogramming. Thus switching from mitochondrial respiration to glycolytic metabolism through regulation of the miR-31/SDHA axis is critical for lowering the reprogramming threshold. This is supportive of multi-stage reprogramming whereby metabolic remodeling is fundamental.
Cancer cells exhibit aerobic glycolysis. This means that cancer cells derive most of their energy from glycolysis that is glucose is converted to lactate for energy followed by lactate fermentation, even when oxygen is available. This is termed the Warburg effect.
Full context extract from my 1998 paper>>>
“So, if mitochondria existed as separate organisms prior to their merging with the drifting Archaea, then it might be expected that they had evolved their own separate aging system. Once the two life forms merged and the larger, combined, life form was completely dependent on the mitochondrial energy source, whenever enough mitochondria in the cell had died, the cell itself would also die. If the mitochondrial imposed death occurred before death caused by telomeric shortening, two aging systems could exist in the same organism, one dominant and one vestigial. Mitochondrial aging will be referred to as Aging System #2 or (AS#2).
The next step in evolution would likely have been the vast colonization of the oceans by these photosynthetic Archaea. (We will now refer to them as algae). With the sun providing unlimited energy and the ocean an unrestricted habitat, evolution would select for maximal reproductive potential and therefore maximal life spans. The first two aging systems, therefore were likely deactivated. The symbiotic mitochondria could simply evolve longer life spans, and the telomeric aging system could be deactivated by the creation of telomerase which rebuilds the ends of the chromosomes after each round of replication. Also, to counter the effect of the sun’s deadly mutating UV, Gamma, and X rays ( referred to herein as solar radiation) a DNA repair system had to evolve that could excise damaged base pairs and replace them with the proper ones. Additionally, to protect against the free radicals generated by the oxygen produced from photosynthesis and solar radiation, an antioxidant protective system had to evolve as well. After a billion years of this selection pressure it could be expected that the algae evolved into non-aging, rapidly-reproducing organisms with perfect DNA repair and free radical defense systems. Is there any evidence that single cell organisms were once immortal? Many cell-types with the proper manipulations can become immortalized cancer strains and reproduce indefinitely as a culture. Cancer, in a broad sense, may simply be a cell returning to its earlier, primitive, immortalized, state. It should not be very surprising that a mortal life form that evolved from a previously immortalized life form could spontaneously become immortalized through loss of some type of control. However, if the mortal life form had evolved from mortal ancestors, spontaneous immortalization would seem to be quite a miracle indeed.
Update #14- A new Horvath article has just been published in Nature..it turns out as expected transient expression of Yamanaka factors can reverse the aging process!!
Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells
Abstract
In my 1998 paper I predicted that antioxidants would be found to catalyze the methylation of cytosines, and free radicals would be found to catalyze the demethylation of cytosines…here is the exact language>>>
This was in the abstract>> “Free radicals catalyze the demethylation of 5mC while antioxidants catalyze the remethylation of cytosine by altering the activity of DNA methyltransferases. Hormones act as either surrogate free radicals by stimulating the cAMP pathway which alters free radical levels within cells, or as surrogate antioxidants through cGMP pathway stimulation. Access to DNA containing 5mC-inhibited developmental and aging genes and restriction sites is allowed by DNA helicase strand separation. Tightly wound DNA does not allow this access. ”
In the concluding remarks>>>
- “A major manner in which genetic signaling occurs is through alteration of the methylation status of 5mC in 5mC demethylation is catalyzed by free radicals and methylation by antioxidants.”
- “The main assumptions that this theory relies upon are that free radicals catalyze the demethylation of 5mC while antioxidants catalyze the methylation of 5mC by influencing DNA methyltransferase activity. This theory was constructed prior to the author’s knowledge that there existed any evidence that supported the above assumptions. After studies were located that confirmed the major assumptions of this theory, the likelihood of it being correct increased significantly.”
So my examination of the 200+ journal articles in Pub Med that contain the term “Yamanaka factors ” yields some very interesting results… It turns out that it appears that the general rule is that adding antioxidants to the Yamanaka factors greatly increases their differentiation into specific cell lines (presumably through increasing general cytosine methylation and shutting down of various genes.). It also appears to be generally true that adding free radicals to the mix greatly increases the loss of differentiation of the cells and allows them to become pluripotent (blank) stem cells (presumably by increasing loss of cytosine methylation) .
Now, if these phenomena are not occurring via DNA methylation/demethylation, then the other idea that might hold water is that antioxidants catalyze the binding of transcription factors to their binding sites while free radicals catalyze the removal of transcription factors. Another way to think of it is that in essence is that methyl groups are the simplest form of transcription factors and the antioxidants and free radicals are catalyzing all the reactions.
Well what do you know? This all seems to be revealed to likely be true in the following relevant abstracts of Yamanaka factor articles. There is one odd abstract however where it refers to a TET protein that is involved with demethylating cytosines and antioxidants increase its activity of demethylation! Here is a definition of TET protein’s. The TET enzymes are a family of ten-eleven translocation (TET) methylcytosine dioxygenases. They are instrumental in DNA demethylation of 5-Methylcytosine.
This little clue gives me an idea as to what to look for to find the methyltransferase that is activated during aging to methylate the 36 development-related genes uncovered by Horvath. There might exist a special methyltransferase that is activated by free radicals to methylate those 36 genes. This would make sense if the pro-aging cAMP stimulating hormones LH, FSH, and hCG are in actuality as I predicted in the 1998 paper – free radical generating hormones.
Okay here are the interesting abstracts>>>
Ascorbic acid promotes the direct conversion of mouse fibroblasts into beating cardiomyocytes
Abstract
Recent advances in the direct conversion of fibroblasts to cardiomyocytes suggest this process as a novel promising approach for cardiac cell-based therapies. Here, by screening the effects of 10 candidate small molecules along with transient overexpression of Yamanaka factors, we show ascorbic acid (AA), also known as vitamin C, enhances reprogramming of mouse fibroblasts into beating cardiomyocytes. Immunostaining and gene expression analyses for pluripotency and cardiac lineage markers confirmed beating patches were derived from non-cardiac lineage cells without passing through a pluripotent intermediate. Further analysis revealed that AA also increased the size of the beating areas and the number of cardiac progenitors. Immunostaining for cardiac markers, as well as electrophysiological analysis confirmed the functionality of directly converted cardiomyocytes. These results illustrate the importance of AA in direct conversion of fibroblasts to cardiomyocytes and may open new insights into future biomedical applications for induced cardiomyocytes.
Optimal ROS Signaling Is Critical for Nuclear Reprogramming
Free PMC article
Abstract
Efficient nuclear reprogramming of somatic cells to pluripotency requires activation of innate immunity. Because innate immune activation triggers reactive oxygen species (ROS) signaling, we sought to determine whether there was a role of ROS signaling in nuclear reprogramming. We examined ROS production during the reprogramming of doxycycline (dox)-inducible mouse embryonic fibroblasts (MEFs) carrying the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc [OSKM]) into induced pluripotent stem cells (iPSCs). ROS generation was substantially increased with the @@@@@@READ THIS- I believe ROS catalyze the demethylation of 5mC!
onset of reprogramming. Depletion of ROS via antioxidants or Nox inhibitors substantially decreased reprogramming efficiency. Similarly, both knockdown and knockout of p22(phox)-a critical subunit of the Nox (1-4) complex-decreased reprogramming efficiency. However, excessive ROS generation using genetic and pharmacological approaches also impaired reprogramming. Overall, our data indicate that ROS signaling is activated early with nuclear reprogramming, and optimal levels of ROS signaling are essential to induce pluripotency.
Free PMC article
Abstract
The ability to efficiently generate integration-free induced pluripotent stem cells (iPSCs) from the most readily available source-peripheral blood-has the potential to expedite the advances of iPSC-based therapies. We have successfully generated integration-free iPSCs from cord blood (CB) CD34(+) cells with improved oriP/EBNA1-based episomal vectors (EV) using a strong spleen focus forming virus (SFFV) long terminal repeat (LTR) promoter. Here we show that Yamanaka factors (OCT4, SOX2, MYC, and KLF4)-expressing EV can also reprogram adult peripheral blood mononuclear cells (PBMNCs) into pluripotency, yet at a very low efficiency. We found that inclusion of BCL-XL increases the reprogramming efficiency by approximately 10-fold. Furthermore, culture of CD3(-)/CD19(-) cells or T/B cell-depleted MNCs for 4-6 days led to the generation of 20-30 iPSC colonies from 1 ml PB, an efficiency that is substantially higher than previously reported. PB iPSCs express pluripotency markers, form teratomas, and can be induced to differentiate in vitro into mesenchymal stem cells, cardiomyocytes, and hepatocytes. Used together, our optimized factor combination and reprogramming strategy lead to efficient generation of integration-free iPSCs from adult PB. This discovery has potential applications in iPSC banking, disease modeling and regenerative medicine.
Cell 1993 Oct 22;75(2):241-51.
Bcl-2 functions in an antioxidant pathway to prevent apoptosis
Abstract
Bcl-2 inhibits most types of apoptotic cell death, implying a common mechanism of lethality. Bcl-2 is localized to intracellular sites of oxygen free radical generation including mitochondria, endoplasmic reticula, and nuclear membranes. Antioxidants that scavenge peroxides, N-acetylcysteine and glutathione peroxidase, countered apoptotic death, while manganese superoxide dismutase did not. Bcl-2 protected cells from H2O2- and menadione-induced oxidative deaths. Bcl-2 did not prevent the cyanide-resistant oxidative burst generated by menadione. Two model systems of apoptosis showed no increment in cyanide-resistant respiration, and generation of endogenous peroxides continued at an inherent rate that was unaltered by Bcl-2. Following an apoptotic signal, cells sustained progressive lipid peroxidation. Overexpression of Bcl-2 functioned to suppress lipid peroxidation completely. We propose a model in which Bcl-2 regulates an antioxidant pathway at sites of free radical generation.
JMJD3 acts in tandem with KLF4 to facilitate reprogramming to pluripotency
Free PMC article
Abstract
The interplay between the Yamanaka factors (OCT4, SOX2, KLF4 and c-MYC) and transcriptional/epigenetic co-regulators in somatic cell reprogramming is incompletely understood. Here, we demonstrate that the histone H3 lysine 27 trimethylation (H3K27me3) demethylase JMJD3 plays conflicting roles in mouse reprogramming. On one side, JMJD3 induces the pro-senescence factor Ink4a and degrades the pluripotency regulator PHF20 in a reprogramming factor-independent manner. On the other side, JMJD3 is specifically recruited by KLF4 to reduce H3K27me3 at both enhancers and promoters of epithelial and pluripotency genes. JMJD3 also promotes enhancer-promoter looping through the cohesin loading factor NIPBL and ultimately transcriptional elongation. This competition of forces can be shifted towards improved reprogramming by using early passage fibroblasts or boosting JMJD3’s catalytic activity with vitamin C. Our work, thus, establishes a multifaceted role for JMJD3, placing it as a key partner of KLF4 and a scaffold that assists chromatin interactions and activates gene transcription.
Nuclear S-Nitrosylation Defines an Optimal Zone for Inducing Pluripotency
Abstract
Background: We found that cell-autonomous innate immune signaling causes global changes in the expression of epigenetic modifiers to facilitate nuclear reprogramming to pluripotency. A role of S-nitrosylation by inducible nitric oxide (NO) synthase, an important effector of innate immunity, has been previously described in the transdifferentiation of fibroblasts to endothelial cells. Accordingly, we hypothesized that S-nitrosylation might also have a role in nuclear reprogramming to pluripotency.
Methods: We used murine embryonic fibroblasts containing a doxycycline-inducible cassette encoding the Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc), and genetic or pharmacological inhibition of inducible NO synthase together with the Tandem Mass Tag approach, chromatin immunoprecipitation-quantitative polymerase chain reaction, site-directed mutagenesis, and micrococcal nuclease assay to determine the role of S-nitrosylation during nuclear reprogramming to pluripotency.
Results: We show that an optimal zone of innate immune activation, as defined by maximal yield of induced pluripotent stem cells, is determined by the degree of activation of nuclear factor κ-light-chain-enhancer of activated B cells; NO generation; S-nitrosylation of nuclear proteins; and DNA accessibility as reflected by histone markings and increased mononucleosome generation in a micrococcal nuclease assay. Genetic or pharmacological inhibition of inducible NO synthase reduces DNA accessibility and suppresses induced pluripotent stem cell generation. (free radicals catalyze demethylation) The effect of NO on DNA accessibility is mediated in part by S-nitrosylation of nuclear proteins, including MTA3 (Metastasis Associated 1 Family Member 3), a subunit of NuRD (Nucleosome Remodeling Deacetylase) complex. S-Nitrosylation of MTA3 is associated with decreased NuRD activity. Overexpression of mutant MTA3, in which the 2 cysteine residues are replaced by alanine residues, impairs the generation of induced pluripotent stem cells.
Conclusions: This is the first report showing that DNA accessibility and induced pluripotent stem cell yield depend on the extent of cell-autonomous innate immune activation and NO generation. This “Goldilocks zone” for inflammatory signaling and epigenetic plasticity may have broader implications for cell fate and phenotypic fluidity.
This one doesn’t fit>>>>
. 2019 Feb 12;5:11.
Free PMC article
Abstract
The relationship between active DNA demethylation induced by overexpressing Tet1 and passive DNA demethylation induced by suppressing Dnmt1 remains unclear. Here, we found that DNMT1 preferentially methylated, but TET1 preferentially demethylated, hemi-methylated CpG sites. These phenomena resulted in a significant overlap in the targets of these two types of DNA demethylation and the counteractions of Dnmt1 and Tet1 during somatic cell reprogramming. Since the hemi-methylated CpG sites generated during cell proliferation were enriched at core pluripotency loci, DNA demethylation induced by Tet1 or sh-RNA against Dnmt1 (sh-Dnmt1) was enriched in these loci, which, in combination with Yamanaka factors, led to the up-regulation of these genes and promoted somatic cell reprogramming. In addition, since sh-Dnmt1 induces DNA demethylation by impairing the further methylation of hemi-methylated CpG sites generated during cell proliferation, while Tet1 induced DNA demethylation by demethylating these hemi-methylated CpG sites, Tet1-induced DNA demethylation, compared with sh-Dnmt1-induced DNA demethylation, exhibited a higher ability to open the chromatin structure and up-regulate gene expression. Thus, Tet1-induced but not sh-Dnmt1-induced DNA demethylation led to the up-regulation of an additional set of genes that can promote the epithelial-mesenchymal transition and impair reprogramming. ????????? When vitamin C was used to further increase the demethylation ability ?????????of TET1 during reprogramming, Tet1 induced a larger up-regulation of these genes and significantly impaired reprogramming. Therefore, the current studies provide additional information regarding DNA demethylation during somatic cell reprogramming.
Stem Cell Res. 2016 Sep;17(2):296-305.
Free article
Abstract
Biallelic mutations in ATM result in the neurodegenerative syndrome Ataxia-Telangiectasia, while ATM haploinsufficiency increases the risk of cancer and other diseases. Previous studies revealed low reprogramming efficiency from A-T and carrier fibroblasts, a barrier to iPS cell-based modeling and regeneration. Here, we tested the feasibility of employing circulating erythroid cells, a compartment no or minimally affected in A-T, for the generation of A-T and carrier iPS cells. Our results indicate that episomal expression of Yamanaka factors plus BCL-xL in erythroid cells results in highly efficient iPS cell production in feeder-free, xeno-free conditions. Moreover, A-T iPS cells generated with this protocol maintain long-term replicative potential, stable karyotypes, re-elongated telomeres and capability to differentiate along the neural lineage in vitro and to form teratomas in vivo. Finally, we find that haploinsufficiency for ATM does not limit reprogramming from human erythroid cells or in vivo teratoma formation in the mouse.
Update #16- in progress
After writing update #15 it quickly became obvious that we need to focus on the activity of the odd TET enzymes that in the presence of antioxidants maintain the unmethylated status of various unmethylated genes. Do they maintain the methylation status of Horvath’s 36 development/aging genes the become hypermethylated (probably gradually) during aging? Does the decline in the antioxidant status of older individuals lead to the turning off of these TET enzymes which allows the methylation of various genes? Why does the antioxidant status of individuals decline with aging? Let’s take another look at the hormones that change the most with aging.
We see a huge decline in the hormones melatonin, DHEA, pregnenolone, estrogen, progesterone, and DHEA. What do all these hormones have in common? They all are either antioxidants (some very strong) or at least stimulate antioxidant activity. We also see a huge rise in a a number of (I propose) free radical stimulating hormones like FSH, LH, and hCG. Here is some info about these hormones:
Melatonin as an antioxidant: under promises but over delivers
Melatonin is uncommonly effective in reducing oxidative stress under a remarkably large number of circumstances. It achieves this action via a variety of means: direct detoxification of reactive oxygen and reactive nitrogen species and indirectly by stimulating antioxidant enzymes while suppressing the activity of pro-oxidant enzymes. In addition to these well-described actions, melatonin also reportedly chelates transition metals, which are involved in the Fenton/Haber-Weiss reactions; in doing so, melatonin reduces the formation of the devastatingly toxic hydroxyl radical resulting in the reduction of oxidative stress. Melatonin’s ubiquitous but unequal intracellular distribution, including its high concentrations in mitochondria, likely aid in its capacity to resist oxidative stress and cellular apoptosis. There is credible evidence to suggest that melatonin should be classified as a mitochondria-targeted antioxidant. Melatonin’s capacity to prevent oxidative damage and the associated physiological debilitation is well documented in numerous experimental ischemia/reperfusion (hypoxia/reoxygenation) studies especially in the brain (stroke) and in the heart (heart attack). Melatonin, via its antiradical mechanisms, also reduces the toxicity of noxious prescription drugs and of methamphetamine, a drug of abuse. Experimental findings also indicate that melatonin renders treatment-resistant cancers sensitive to various therapeutic agents and may be useful, due to its multiple antioxidant actions, in especially delaying and perhaps treating a variety of age-related diseases and dehumanizing conditions. Melatonin has been effectively used to combat oxidative stress, inflammation and cellular apoptosis and to restore tissue function in a number of human trials; its efficacy supports its more extensive use in a wider variety of human studies. The uncommonly high-safety profile of melatonin also bolsters this conclusion. It is the current feeling of the authors that, in view of the widely diverse beneficial functions that have been reported for melatonin, these may be merely epiphenomena of the more fundamental, yet-to-be identified basic action(s) of this ancient molecule.
Front Pharmacol. 2020; 11: 360.
Pregnenolone Inhibits Osteoclast Differentiation and Protects Against Lipopolysaccharide-Induced Inflammatory Bone Destruction and Ovariectomy-Induced Bone Loss
Antioxidant effects of dehydroepiandrosterone (DHEA) and 7alpha-hydroxy-dehydroepiandrosterone in the rat colon, intestine and liver
This study examined in healthy male Wistar rats the in vivo antioxidant effect of dehydroepiandrosterone (DHEA) and 7alpha-hydroxy-DHEA administered by intraperitoneal injections (50 mg/kg body weight) for 2 or 7 days. Markers of oxidative damage to lipids (thiobarbituric acid-reacting substances, TBARS) and to proteins (protein carbonyls) were assessed in colon, small intestine, and liver homogenates. DHEA and 7alpha-hydroxy-DHEA caused a decrease in body weight. DHEA treatment significantly increased liver, colon, and small intestine cell weights. After 7 days, DHEA exerted an antioxidant effect in all organs studied. In the colon, oxidative damage protection was accompanied by a goblet cell proliferation and increase in acidic mucus production. After 2 days, the antioxidant effect of 7alpha-hydroxy-DHEA was mainly observed in the liver. Nonprotein sulfhydryl groups (mostly glutathione levels) were altered by DHEA in the liver whereas they remained unchanged after 7alpha-hydroxy-DHEA treatment. The results indicate that in healthy animals, DHEA exerts a protective effect, particularly in the colon, by reducing the tissue susceptibility to oxidation of both lipids and proteins. This effect was not limited to a specific tissue, whereas the metabolite 7alpha-hydroxy-DHEA exerted its antioxidant effect towards the two markers of oxidative damage earlier than DHEA, and mainly in the liver.
Spotlight on a New Heme Oxygenase Pathway: Testosterone-Induced Shifts in Cardiac Oxidant/Antioxidant Status
Human chorionic gonadotropin (hCG) may be a marker of systemic oxidative stress in normotensive and preeclamptic term pregnancies
In vitro studies on placental function have revealed interactions between levels of secretion of human chorionic gonadotropin (hCG) by trophoblastic cells and oxidative stress generated by hydrogen peroxide (H2O2). Here, we have examined the relationship between maternal levels of hCG and H2O2 in vivo in term pregnancies with and without preeclampsia. We measured serum levels of hCG and H2O2 in twenty preeclamptic and twenty normotensive term pregnant women (controls), using an enzymatic immunoassay and an electrochemical method, respectively. Higher levels of serum hCG and H2O2 were observed in patients with preeclampsia in comparison to controls. A significant positive correlation between serum hCG concentration and H2O2 production was found. Our results show that: (1) systemic hCG levels are correlated with an oxidative stress state in term pregnant women with preeclampsia and (2) circulating hCG may be a monitoring tool of oxidative stress during pregnancy.
FSH is an odd case and I have yet to completely unravel its mystery. Given that it is a cAMP stimulating hormone like LH and hCG I would expect it to be primarily a free radical generating hormone. However FSH also has antioxidant like qualities. One case is that just like antioxidants, FSH can protect the developing ovum from free radical damage. But this hormone increases dramatically after age 50 in both men and women. I have also found that FSH is not associated with any cancers when I did a Pub Med search on this question for my 1998 paper. However, all the other cAMP stimulating hormones were in some way associated with various cancers. I believe somehow FSH is associated with the development/aging program that is accelerated in progeria as kids with progeria never get cancer. Somehow FSH must be associated with Lamin A proteins- this would be a good area for some studies. Maybe FSH is like estrogen and progesterone having both free radical and antioxidant potential.
FSH protects mouse granulosa cells from oxidative damage by repressing mitophagy
Scientific Reports volume 6, Article number: 38090 (2016)
Oxidative stress has been implicated in triggering granulosa cell (GC) death during follicular atresia. Recent studies suggested that follicle-stimulating hormone (FSH) has a pivotal role in protecting GCs from oxidative injury, although the exact mechanism remains largely unknown. Here, we report that FSH promotes GC survival by inhibiting oxidative stress-induced mitophagy. The loss of GC viability caused by oxidative stress was significantly reduced after FSH treatment, which was correlated with impaired activation of mitophagy upon oxidative stress. Compared with FSH treatment, blocking mitophagy displayed approximate preventive effect on oxidative stress-induced GC death, but FSH did not further restore viability of cells pretreated with mitophagy inhibitor. Importantly, FSH suppressed the induction of serine/threonine kinase PINK1 during oxidative stress. This inhibited the mitochondrial translocation of the E3 ligase Parkin, which is required for the subsequent clearance of mitochondria, and ultimately cell death via mitophagy. In addition, knocking down PINK1 using RNAi confirmed the role of the FSH-PINK1-Parkin-mitophagy pathway in regulating GC survival under oxidative conditions. These findings introduce a novel physiological function of FSH in protecting GCs against oxidative damage by targeting PINK1-Parkin-mediated mitophagy.
My 1998 paper predicted that hormones that stimulate the cAMP pathway could be considered free radical hormones and hormones that stimulated the cGMP pathway could be considered antioxidant hormones :
cGMP Signaling Increases Antioxidant Gene Expression by Activating Forkhead Box O3A in the Colon Epithelium
Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway
Abstracts concerning TET enzymes>>>
Analysis of the machinery and intermediates of the 5hmC-mediated DNA demethylation pathway in aging on samples from the MARK-AGE Study- Published 2016
Abstract
Gradual changes in the DNA methylation landscape occur throughout aging virtually in all human tissues. A widespread reduction of 5-methylcytosine (5mC), associated with highly reproducible site-specific hypermethylation, characterizes the genome in aging. Therefore, an equilibrium seems to exist between general and directional deregulating events concerning DNA methylation controllers, which may underpin the age-related epigenetic changes. In this context, 5mC-hydroxylases (TET enzymes) are new potential players. In fact, TETs catalyze the stepwise oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), driving the DNA demethylation process based on thymine DNA glycosylase (TDG)-mediated DNA repair pathway. The present paper reports the expression of DNA hydroxymethylation components, the levels of 5hmC and of its derivatives in peripheral blood mononuclear cells of age-stratified donors recruited in several European countries in the context of the EU Project ‘MARK-AGE’. The results provide evidence for an age-related decline of TET1, TET3 and TDG gene expression along with a decrease of 5hmC and an accumulation of 5caC. These associations were independent of confounding variables, including recruitment center, gender and leukocyte composition. The observed impairment of 5hmC-mediated DNA demethylation pathway in blood cells may lead to aberrant transcriptional programs in the elderly.
This Looks Interesting-
Tet1 Deficiency Leads to Premature
Reproductive Aging by Reducing Spermatogonia
Stem Cells and Germ Cell Differentiation
SUMMARY
Ten-eleven translocation (Tet) enzymes are involved in DNA demethylation, important in regulating
embryo development, stem cell pluripotency and tumorigenesis. Alterations of DNA methylation
with age have been shown in various somatic cell types. We investigated whether Tet1 and Tet2 regulate aging. We showed that Tet1-deficient mice undergo a progressive reduction of spermatogonia
stem cells and spermatogenesis and thus accelerated infertility with age. Tet1 deficiency decreases
5hmC levels in spermatogonia and downregulates a subset of genes important for cell cycle, germ
cell differentiation, meiosis and reproduction, such as Ccna1 and Spo11, resulting in premature reproductive aging. Moreover, Tet1 and 5hmC both regulate signaling pathways key for stem cell development, including Wnt and PI3K-Akt, autophagy and stress response genes. In contrast, effect of Tet2
deficiency on male reproductive aging is minor. Hence, Tet1 maintains spermatogonia stem cells
with age, revealing an important role of Tet1 in regulating stem cell aging.
Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus
Abstract
Purpose
Aging is believed to affect epigenetic marking of brain DNA with 5-methylcytosine (5mC) and possibly via the 5mC to 5-hydroxymethylcytosine (5hmC) conversion by TET (ten-eleven translocation) enzymes. We investigated the impact of aging on hippocampal DNA 5-hydroxymethylation including in the sequence of aging-susceptible 5-lipoxygenase (5-LOX) gene.
Methods
Hippocampal samples were obtained from C57BL6 mice. Cellular 5hmC localization was determined by immunofluorescence. The global 5mC and 5hmC contents were measured with the corresponding ELISA. The 5-LOX 5hmC content was measured using a glucosyltransferase/enzymatic restriction digest assay. TET mRNA was measured using qRT-PCR.
Results
Global hippocampal 5hmC content increased during aging as did the 5hmC content in the 5-LOX gene. This occurred without alterations of TET1–3 mRNAs and without changes in the content of 8-hydroxy-2-deoxy-guanosine, a marker of non-enzymatic DNA oxidation.
Conclusions
The aging-associated increase of hippocampal 5hmC content (global and 5-LOX) appears to be unrelated to oxidative stress. It may be driven by an altered activity but not by the increased expression of the three TET enzymes. Global 5hmC content was increased during aging in the absence of 5mC decrease, suggesting that 5hmC could act as an epigenetic marker and not only as an intermediary in DNA demethylation. Further research is needed to elucidate the functional implications of the impact of aging on hippocampal cytosine hydroxymethylation.
Additional abstracts of interest>>>
Update #17 Caloric Restriction (in progress)
During caloric restriction we find that antioxidant hormones increase dramatically while free radical hormones decline dramatically-this leads to an altered (increased) antioxidant status in the nucleoplasm. This in turn would lead to the TET enzymes working better at keeping the 36 anti-aging genes demethylated. This should lead to rejuvenating the animal undergoing CR at at least slowing the aging process. This allows the aging system to be malleable depending on the hormonal milieu which is dictated by environmental conditions. It even allows for reversal of aging symptoms which would be beneficial during a famine for making sure that if a single mating pair of individuals survived a long famine (or drought) they would be young enough to reproduce and reconstitute the group…SO now we can see why melatonin given to women who have recently entered menopause causes them to experience menopause reversal.
In my 1998 paper I studied the hormone changes that occur during caloric restriction and came up with this from the paper
“Melatonin: the famine and drought hormone.
During famine conditions or CR one would expect that in addition to the increase in cGMP activity, that an increase in cGMP stimulating hormones would be seen. Also, one would expect a decline in cAMP stimulating hormones. In a study of human males undergoing 5 days of fasting (136) the following hormone level changes were seen, (for hormones not measured in this study other references are noted):
cAMP stimulating hormones:
TSH declined by 67%-as expected
LH decreased by 33%-as expected
FSH decreased by 33%-as expected
cortisol increased by 110%-unexpected
estrogen -increased by 10%-unexpected
cGMP stimulating hormones
Melatonin increased +/-100% in rats (137)-as expected
GH increased 200%-400% in men (138) -as expected
DHEA-S increased 100%-expected
Testosterone-decreased 50%- unexpected
T3 and T4 were relatively unaffected, and prolactin declined 25% but is not listed because it is an “ambidextrous” hormone stimulating both cAMP and cGMP depending on which receptors it influences.
The above results reasonably conform to expectations based on the prior hypothesis regarding cAMP and cGMP stimulating hormones. However, by examining the exceptions additional important insights can be gained. First, the cortisol increase of 110% is definitely not expected as it is a cAMP hormone and the hormone is widely known to be implicated in accelerating the diseases of aging in persons where it is chronically elevated. What is also known about cortisol is that it has been implicated in triggering apoptosis is various cell types including thymocytes of the thymus gland (139). If the early stages of CR require a large scale induction of apoptosis in various cells, it is likely that the increased cortisol is involved. The other contradiction about the large cortisol increase is that when it occurs during CR one must assume that it does not lead to the deleterious accelerated age changes that are normally associated with high cortisol levels as CR’d animals live much longer than controls. One study explains the contradiction: during CR, although the baseline levels of cortisol are elevated, increases in peak cortisol levels from stress are shown to be lower in CR’d animals than in ad lib fed animals (140). The idea that evolution has designed stress so that at times it kills and at other times it does not suggests that stress is also an aging system. This idea will be explored shortly.
The other exceptions include a 10% increase in estrogen and a 50% decrease in testosterone. If one remembers that inhibition of reproduction would be a primary goal of the CR response, then a drop in the male reproductive hormone is not illogical even though it is a cGMP hormone. The corresponding increase in DHEA of 100% which in absolute terms is of equal magnitude to the testosterone decline might be seen as CR’s version of testosterone that does not induce sexuality in the male. Finally, if the only male aging symptoms associated with AS#6 include prostatic atrophy (assuming no malfunctions in apoptosis) then the estrogen increase of 10% can also be seen as an anti-reproductive hormone change. An estrogen increase however, would not be expected to occur in the female during CR, and studies show that this is likely true (141).
CR leads to quite a complicated array of hormone changes, but can it all be simplified? A simple Medline search of melatonin against each of the individual hormones mentioned above provides the answer. Melatonin administration has been shown to suppress LH (142), FSH (143), and testosterone(144) while increasing DHEA(145), GH(146), and in some cases cortisol(147) levels in either rats, mice or humans. In females, 300 mg. of melatonin was shown to suppress estrogen (E2) levels (148). More definitive studies do need to be made in this area, however, as most studies are short term in nature while melatonin induced hormone changes seem to take much longer to occur in humans. Melatonin’s effect on prolactin, however, was not clear and is generally suggestive of increasing levels in humans but this might only be a short term effect due to the short term nature of human melatonin studies. Melatonin, did however, reduce prolactin levels in the rat pituitary (149). TSH was also shown to be suppressed in the rat by melatonin (150) In most cases of hormone changes induced by CR, melatonin administration induced the same effect. What is also interesting, a reduction in body temperature in animals is seen during CR and posited by some to be the potential candidate as the active life-extending mechanism in CR. As one would expect, melatonin administration leads to reduced body temperature as well (151a). It is interesting to note that water deprivation, as would be expected, has also been shown to increase melatonin levels in rodents (151b).”
One other thing I noticed long ago was that during fasting or CR, Beta Hydroxy Butyrate (BHB) increases by 1,000%+. For a long time I ignored this thinking it was just a result of the body burning fat for fuel instead of consuming calories. But not too long ago I searched BHB vs aging and other things and found BHB has numerous anti aging and pro-health effects, including helping to treat colorectal cancer!
The #18 The Wall of Shame:
A List of Aging Researchers Who
Laughed The Hardest at the Idea of Programmed Aging
Michael Lemonick (editor of the much diminished Scientific American)
Michael Fossel (MD)
Stephen Spindler
Richard Miller
Arlan Richardson
Gordon Lithgow
Leonard Hayflick
Aubrey De Grey
Brian Charlesworth
Marc Tatar
Update #19
I always was curious about this study and now I think we have shed some light on it. BHT certainly looks like a molecule that could colocalize with DNA.
Effects of the antioxidant butylated hydroxytoluene (BHT) on mortality in BALB/c mice
Butylated hydroxytoluene (BHT) was given in the feed to determine its effect on life span in genetically well-defined, barrier-derived BALB/c mice. Both sexes received 0.75% BHT for three different treatment periods: (A) 8 to 11 weeks of age; (B) for life, beginning at 11 weeks; (C) for life, beginning at 8 weeks of age. The control group (D) was untreated. All BHT treatment groups had mean survival times which exceeded that of controls. The order of survival was B greater than C greater than A greater than D (Males: 890, 832, 726, 684 days; Females: 875, 798, 759, 701 days). Most of the increases in mean survival time were related to a reduction in early deaths (350–600 days) in BHT-treated mice. The reason for the life-lengthening effect on BHT was not identified, but it may relate to alterations in specific disease incidences.

Update #20
Vitamin D3 is also a hormone with antioxidant effects that declines with age. Why does it decline? Elderly skin makes less than half the amount of Vitamin D3 as young skin does from the same amount of sunlight.
Abstract
In healthy individuals brain iron levels increase with age (Bartzokis et al., 1994) and abnormally high brain iron levels are observed in age-related degenerative diseases.)
Here is an interesting abstract>>>
The Aging of Iron Man
Brain iron is tightly regulated by a multitude of proteins to ensure homeostasis. Iron dyshomeostasis has become a molecular signature associated with aging which is accompanied by progressive decline in cognitive processes. A common theme in neurodegenerative diseases where age is the major risk factor, iron dyshomeostasis coincides with neuroinflammation, abnormal protein aggregation, neurodegeneration, and neurobehavioral deficits. There is a great need to determine the mechanisms governing perturbations in iron metabolism, in particular to distinguish between physiological and pathological aging to generate fruitful therapeutic targets for neurodegenerative diseases. Based on the evidence discussed here, we suggest a synergistic use of iron-chelators and anti-inflammatories as putative anti-brain aging therapies to counteract pathological aging in neurodegenerative diseases.
Update #21
After pondering the idea that the accumulation of iron in the elderly participates in driving the aging process by increasing the oxidation potential of the cellular environment, I then wondered if deficiencies in other minerals that are very common in the elderly also helped drive the aging process by reducing the antioxidant potential in the cell…There are two critical factors that are very often deficient in the elderly due to increasing difficulties in absorbing these minerals from the diet as one ages. It is estimated that 80% to 95% of the elderly are magnesium deficient. Most are zinc deficient as well.
Magnesium in Aging, Health and Diseases
Abstract: Several changes of magnesium (Mg) metabolism have been reported with aging, including
diminished Mg intake, impaired intestinal Mg absorption and renal Mg wasting. Mild Mg deficits
are generally asymptomatic and clinical signs are usually non-specific or absent. Asthenia, sleep
disorders, hyperemotionality, and cognitive disorders are common in the elderly with mild Mg
deficit, and may be often confused with age-related symptoms. Chronic Mg deficits increase the
production of free radicals which have been implicated in the development of several chronic age-related disorders. Numerous human diseases have been associated with Mg deficits, including
cardiovascular diseases, hypertension and stroke, cardio-metabolic syndrome and type 2 diabetes
mellitus, airways constrictive syndromes and asthma, depression, stress-related conditions and
psychiatric disorders, Alzheimer’s disease (AD) and other dementia syndromes, muscular diseases
(muscle pain, chronic fatigue, and fibromyalgia), bone fragility, and cancer. Dietary Mg and/or Mg
consumed in drinking water (generally more bioavailable than Mg contained in food) or in alternative
Mg supplements should be taken into consideration in the correction of Mg deficits. Maintaining
an optimal Mg balance all through life may help in the prevention of oxidative stress and chronic
conditions associated with aging. This needs to be demonstrated by future studies.
Zinc, aging, and immunosenescence: an overview
Abstract
Zinc plays an essential role in many biochemical pathways and participates in several cell functions, including the immune response. This review describes the role of zinc in human health, aging, and immunosenescence. Zinc deficiency is frequent in the elderly and leads to changes similar to those that occur in oxidative inflammatory aging (oxi-inflamm-aging) and immunosenescence. The possible benefits of zinc supplementation to enhance immune function are discussed.
Magnesium increases the activity of an important glutathione enzyme called glutathione peroxidase (GPx). This enzyme accelerates how quickly glutathione neutralizes free radicals. Because free radicals are often considered to be “Public Enemy Number One” when it comes to cellular health, longevity and quality of life, you want antioxidants to neutralize them and protect against their damaging effects as quickly as possible. Supplementation with magnesium wasn’t just relegated to glutathione; it also increased the activity of two other extremely important antioxidants that reside in our bodies—catalase and superoxide dismutase.
Zinc is an Antioxidant and Anti-Inflammatory Agent: Its Role in Human Health
Zinc supplementation trials in the elderly showed that the incidence of infections was decreased by approximately 66% in the zinc group. Zinc supplementation also decreased oxidative stress biomarkers and decreased inflammatory cytokines in the elderly. In our studies in the experimental model of zinc deficiency in humans, we showed that zinc deficiency per se increased the generation of IL-1β and its mRNA in human mononuclear cells following LPS stimulation. Zinc supplementation upregulated A20, a zinc transcription factor, which inhibited the activation of NF-κB, resulting in decreased generation of inflammatory cytokines. Oxidative stress and chronic inflammation are important contributing factors for several chronic diseases attributed to aging, such as atherosclerosis and related cardiac disorders, cancer, neurodegeneration, immunologic disorders and the aging process itself. Zinc is very effective in decreasing reactive oxygen species (ROS). In this review, the mechanism of zinc actions on oxidative stress and generation of inflammatory cytokines and its impact on health in humans will be presented.
Update #22
We are looking for up to 30 people to participate in a demonstration project where they will get their DNA methylation age tested and then embark on a 3 to 4 month protocol involving taking high dose melatonin (up to 400 mg/night) , DHEA, pregnenolone, and Vitamin D3 and then retesting their DNA methylation age to see if, as predicted, the DNA methylation age will be significantly lowered. If interested email me at Jeffbo@aol.com.
Update #23
Just in case you think this is all a lot of theory with no proof, take a look at another Horvath recent study that is in press where he has reversed aging in old rats by 50 to 75%! By using the transient expression of Yamanaka Factors (possibly-the factors have not been disclosed at this point) as designed by Dr. Harold Katcher.
Link>>>> https://www.biorxiv.org/content/10.1101/2020.05.07.082917v1
Reversing age: dual species measurement of epigenetic age with a single clock
Abstract:
Abstract
Young blood plasma is known to confer beneficial effects on various organs in mice. However, it was not known whether young plasma rejuvenates cells and tissues at the epigenetic level; whether it alters the epigenetic clock, which is a highly-accurate molecular biomarker of aging. To address this question, we developed and validated six different epigenetic clocks for rat tissues that are based on DNA methylation values derived from n=593 tissue samples. As indicated by their respective names, the rat pan-tissue clock can be applied to DNA methylation profiles from all rat tissues, while the rat brain-, liver-, and blood clocks apply to the corresponding tissue types. We also developed two epigenetic clocks that apply to both human and rat tissues by adding n=850 human tissue samples to the training data. We employed these six clocks to investigate the rejuvenation effects of a plasma fraction treatment in different rat tissues. The treatment more than halved the epigenetic ages of blood, heart, and liver tissue. A less pronounced, but statistically significant, rejuvenation effect could be observed in the hypothalamus. The treatment was accompanied by progressive improvement in the function of these organs as ascertained through numerous biochemical/physiological biomarkers and behavioral responses to assess cognitive functions. Cellular senescence, which is not associated with epigenetic aging, was also considerably reduced in vital organs. Overall, this study demonstrates that a plasma-derived treatment markedly reverses aging according to epigenetic clocks and benchmark biomarkers of aging.
Update #24 Great News!
Another antioxidant that declines dramatically with age (some say 90%) , but at least 50% has been identified >> Alpha -Keto-Glutarate . Which just so happens, when combined with Vitamin C, keeps the TET enzymes working that keep your 36 anti aging genes turned on by demethylating them. Studies also show that this antioxidant extends the lifespan of mice by about 15% and increases health span as well. Also, a related antioxidant that increases by 1,000% during caloric restriction Butylated Hydroxy Butyrate has been shown to have many health improving and anti aging effects and has recently been shown effective in treating colorectal cancer!!
BHB
Update #25 More Great News!
Aging Cell. 2019 Dec; 18(6): e13028.
Epigenetic “clocks” can now surpass chronological age in accuracy for estimating biological age. Here, we use four such age estimators to show that epigenetic aging can be reversed in humans. Using a protocol intended to regenerate the thymus ( DHEA, Growth Hormone, and metformin adminstration), we observed protective immunological changes, improved risk indices for many age‐related diseases, and a mean epigenetic age approximately 1.5 years less than baseline after 1 year of treatment (−2.5‐year change compared to no treatment at the end of the study). The rate of epigenetic aging reversal relative to chronological age accelerated from −1.6 year/year from 0–9 month to −6.5 year/year from 9–12 month. The GrimAge predictor of human morbidity and mortality showed a 2‐year decrease in epigenetic vs. chronological age that persisted six months after discontinuing treatment. This is to our knowledge the first report of an increase, based on an epigenetic age estimator, in predicted human lifespan by means of a currently accessible aging intervention.
Update #26 More Great News!
7 months of Ca-AKG supplementation in humans reversed DNA methylation age by 8 years!
Priority Research Paper Volume 13, Issue 22 pp 24485—24499
Rejuvant®, a potential life-extending compound formulation with alpha-ketoglutarate and vitamins, conferred an average 8 year reduction in biological aging, after an average of 7 months of use, in the TruAge DNA methylation test
Abstract
The search continues for possible interventions that delay and/or reverse biological aging, resulting in extended healthspan and lifespan. Interventions delaying aging in animal models are well established; however, most lack validation in humans. The length of human lifespan makes it impractical to perform survival analysis. Instead, aging biomarkers, such as DNA methylation (DNAm) clocks, have been developed to monitor biological age. Herein we report a retrospective analysis of DNA methylation age in 42 individuals taking Rejuvant®, an alpha-ketoglutarate based formulation, for an average period of 7 months. DNAm testing was performed at baseline and by the end of treatment with Rejuvant® supplementation. Remarkably, individuals showed an average decrease in biological aging of 8 years (p-value=6.538×10-12). Furthermore, the supplementation with Rejuvant® is robust to individual differences, as indicated by the fact that a large majority of participants decreased their biological age. Moreover, we found that Rejuvant® is of additional benefit to chronologically and biologically older individuals. While continued testing, particularly in a placebo-controlled design, is required, the nearly 8-year reversal in the biological age of individuals taking Rejuvant® for 4 to 10 months is noteworthy, making the natural product cocktail an intriguing candidate to affect human aging.
Update #27 Interesting Story- Amazing Idea- and Funny
The anti aging compounds NAD+ and AKG both play major roles in the Krebs Citric Acid Cycle- I think this might be the original primordial aging system!
Update #29 CD38 destroys NAD+ and increases dramatically with age so if you take NAD+ you should also take Luteolin or Apigenin or Fisetin
What is CD38? An Exploration of NAD+ Consumption Uses and …
- Take Taxifolin (diydroquercetin from Siberian larch tree) 50 mg daily.
- Take Apigenin (derived from grapefruit) 50 mg daily.
- Take Luteolin 50 mg daily.
- Eat anthocyanin-rich superfoods, including purple corn extract, pomegranate juice, purple grapes, blackberries, and red…
Bioorganic & Medicinal Chemistry Letters
Flavonoids as inhibitors of human CD38
Abstract
Graphical abstract
Update #30 Putting All the Pieces Together (in progress)-
I am gradually coming to the view that this whole complicated mess can be simplified in the following way.
I believe the 36 Horvath anti-aging genes that get turned off with aging and can be turned on again by taking AKG and Vitamin C to activate one’s TET enzymes. I believe these 36 genes belong to the more ancient somatic development/aging system. I believe these genes are somehow involved with or associated with the accelerated aging disease of progeria.
This leaves the 12 Horvath aging genes that get turned on with aging via demethylation. One of these genes is involved with causing Alzheimer’s disease which is the clue I needed to predict that these 12 genes belong to the sexual development/aging system and thus are likely controlled by the changes in reproduction-related hormones that occur with aging such as increasing LH, FSH. hCG, and declining melatonin, pregnenolone, progesterone, and DHEA (testosterone and estrogen are oddballs so I don’t include them here). One of the 12 genes being associated with Alzheimer’s disease is a clue that points in this direction because it has been shown that elevated LH levels are linked to Alzheimer’s (at least in women)-thus you can call Alzheimer’s a sexual development/aging related disease. And finally, I believe these 12 genes are strongly associated with the accelerated aging disease of Werner’s Syndrome which causes rapid aging only after puberty. However, because Werner’s Syndrome eventually accelerates all types of aging including the rapid aging symptoms seen in progeria, and the mitochondrial rapid aging diseases as well as Ataxia Telangiectasia, Cockayne Syndrome, and Xeroderma Pigmentosum, one can expect that somehow the aging system accelerated in Werner’s Syndrome coopts all these other development/aging programs. Somehow the 12 genes that get turned on with aging , coopt the aging system that turns off the 36 anti aging genes.
Update #31 How Does The Sexual Aging System Co-Opt The Somatic Aging System? –
It just hit me! How Does the Sexual (Werner’s -related) Aging system of the 12 genes that get turned with aging co-opt all the aging symptoms of the somatic aging (progeria-related) system of the 36 genes that get turned off with aging? Remember that one of the 12 genes that gets turned on and is the most expressed aging gene of the 12 genes in all tissues is the LARP1 gene? What was unique about LARP1 protein?
It has an RNA binding site to bind to RNA transcripts. I hypothesized that LARP1 is truncating mRNA transcripts in inappropriate places and is probably the protein that is truncating the Lamin A protein mRNA transcript that is both defective in progeria and in normally aging human cells! This mRNA truncation leads to defective , truncated proteins.
So while the 36 genes that make transcription factors in the somatic aging system are gradually getting shut down as AKG levels decline and the redox state of the cell becomes more free radicalized, the LARP1 protein is accelerating the silencing of the 36 anti aging genes by truncating the mRNA transcripts that are being produced by these 36 genes leading to defective proteins in the event that they are transcribed . LARP1 seems to be a redundant and more insidious tool of aging when compared to aging caused by deactivation of TET enzymes due to declines in AKG. Additionally LARP1 might even be inappropriately truncating an even wider range of mRNAs from other genes outside of the 36 Horvath anti-aging genes. These ideas suggest many new areas of research to pursue. What would be of particular interest would be to discover and categorize all the defective transcription factors that get produced in normally aging individuals. We know of one so far- the Lamin A protein.
Update #32 Why Do The 36 Anti-Aging Genes Have on Average 100 Methylated Cytosines Per Gene, While The 12 Aging Genes Only Have One Methylated Cytosine Each?
Horvath found it interesting that of the 36 genes that get turned off during aging, they each had on average about 100 cytosine methylation sites (3,617 total) . While the 12 genes that become hypomethylated with aging only had 1 cytosine methylation site each. What does this imply?
For the 36 genes that get turned off with aging , having 100 cytosine methylation sites per gene suggests that this aging process is a gradual , stochastic process, that leads to the slow shut down of the gene in question over time.
Having just one methylation site per gene in the 12 aging genes suggests that this aging system at times has to be less gradual and more precise- like human menopause and menopause-related aging. However now that women can live long past the age of menopause, apparently parts of the system have acquired a slower more gradual pace. So how can this aging sytem become gradual even though each gene has only one cytosine methylation site?
It can become a gradual aging system if it is controlled by reproduction/ development /sex-related hormones of FSH, LH, hCG, and the various steroids whose levels change with age like DHEA, pregnenolone, progesterone, and melatonin. Because all these hormones change dramtically but mostly gradually over a lifetime this aging system with the 12 mono-CpG genes does not need any extra cytosine methylation sites. I expect that it is the hormonal milieu that controls whether these genes become demethylated or methylated. It is also likely that aging related demethylation of these genes is not irreversible. I say this because it has been shown that women who have just entered menopause can reverse it and start cycling aging by simply taking melatonin.
Update #33 Just ran a simple pub med search of the 4 Yamanaka factors vs the 4 tissue types /rapid aging diseases in my aging table and got this
OCT4
(+ some c-myc)
Discussion Points
-The only GRC conference I attended was in 1997 it was the 2nd one they had about epigenetics- my submitted abstract published in the conference manual was how DNA methylation controlled aging- it got a lot of laughs.
-Before we start, let me tell you a funny story that is very important-By age 23 I had earned my MBA from Northwestern and passed the CPA exam and started working for a real estate developer. I quit after 1 year and started my own company. But in my mid 20’s I started going to school again to study BioChem to try and find a cure for aging. At age 27 I was back in school! in Bio 101/ I took one class a semester so I could also study aging on the side and could master the material of the class. I was also working in real estate at a reduced level. I set the curve in almost every class even while competing against the premeds. After a number of years, I had completed most of the requirements for a biochem degree. I had finished Bio Chem 101 and was sitting in class in Biochem 201 when they started making us memorize the Krebs citric acid cycle..I just could not take it anymore I figured I wasn’t learning anything important!! So, I dropped out and never finished my degree and then moved over to the Northwestern Med School library to hunker down for about 10 years just reading aging related abstracts. BOY WAS I STUPID!!! TAKE A LOOK AT THE KREBS CITRIC ACID CYCLE!!>>>>>>
It turns out that within the cycle are the intermediates and factors NAD+ and AKG which both decline a lot with age! As we will see they both are potent anti-aging molecules. AKG and vitamin C (ascorbic acid) supplementation will activate the TET enzymes which will keep the 36 Horvath anti-aging genes demethylated. NAD+ supplementation acts to reverse/prevent aging changes associated with chromatin/associated aging and the expression, I believe, of the 12 Horvath pro-aging genes. While humans only make citric acid with this cycle almsot all other animals are making Vitamin C here. Of course, other factors in this cycle like pyruvate or fumarate, malate etc. should be checked for anti-aging activity. Also, not shown here, but BHB is associated with this cycle and is found to increase dramatically during caloric restriction and has all sorts of pro-health and anti-aging effects. This Krebs/Citric Acid Cycle is what makes power in our mitochondria- Is this the primordial aging system that controls the other subsequently evolved aging systems?
-Recently my aging theory has been clarified, due to Horvath’s study on mammalian aging, and from my recent discussions on Mitteldorf’s aging blog about the life extending effects of castration.
-Although I have been aware of these facts for 30+ years, I never really dwelled on the question – what causes an animal to age after castration? A few examples (OF MANY) of animals getting large increases in life span from castration include:
-The Pacific Salmon: their 3-year life span can increase up to 10 years.
-Korean court- eunuchs supposedly live 15 more years on average
-Steve did studies with male sheep showing they can live 60% longer with castration, and showed a slower DNA methylation aging rate.
While my original aging theory paper from 1998 tried to identify 6 different aging systems/programs.. My new thinking is that it can be simplified to just two aging systems…
–An active sex-related aging system driven by changes in hormones and NAD+, &
–An asexual passive aging system driven by loss of differentiation of cells via changes in AKG levels.
A problem with my 1998 paper was that it was too complicated for the typical reader. Most people did not put the effort in needed to understand it I think that includes some of my programmed aging colleagues. Anyway, it was my fault not yours. I made the paper too complicated for anyone to understand without a lot of effort.
Predictions from the 1998 paper that eventually proved true-
-1. Aging is controlled by the same mechanisms that control development.
-2. Dramatic increases in the development/reproduction hormones LH and FSH (and hCG) after age 50 are involved in driving the aging process- the #1 protein that increases with age is derived form LH, the #2 protein is FSH derived.
-3. LH drives neurodegeneration and Alzheimer’s- proved true in a 2005 paper from the NIH
-4. Aging is controlled by DNA methylation (the paper got the 12 genes of Horvath that get turned on with aging/demethylation correct- but just did not imagine the Horvath 36 genes that get turned off with aging by methylation)
-5. The paper correctly predicted that most DNA methyltransferases are catalyzed to methylate cytosines in the presence of antioxidants but demethylate them in the presence of free radicals. (Did not imagine the existence of TET enzymes that work in reverse).
-6. At the time of publication most people believed that telomere shortening caused progeria, the paper predicted it was just a side effect of progeria.
-7. This was an easy one- the paper predicted that due to the successful cloning of Dolly the sheep – that aging was reversible!
Bottom line- the paper was very successful in making many ODD predictions that panned out, suggesting it was on the right track (there were more but the above suffice to make the point).
OK, so now let’s use the results from Horvath’s new study on mammalian aging and try to marry them to elements of my aging theory to see if we can finish this Rubik’s cube!
Horvath found aging causes 12 genes to get turned and 36 genes to get turned off. I believe that the 36 differentiation/development genes that get turned off represent the first aging system to evolve in asexual organisms of gradual loss of cellular differentiation. These 36 genes, I believe become shut down due to loss of activity of TET enzymes due to declining AKG levels with age (there may be other enzymes that operate in reverse but I have not heard of any new candidates). The 12 genes that get turned on with aging represent the sex/reproduction related aging system that evolved with sexual reproduction and I expect are controlled by the changes in sex /development/reproduction related hormones and NAD+ levels with age.
My paper suggested that the 2 main (human) aging systems were seen in an accelerated form in progeria and in Werner’s Syndrome. I present a modified table of accelerated aging symptoms seen in both of these diseases: (it differs a bit from the one in my 1998 paper-it is more simplified)
(I am keeping it simple with just 2 development/aging systems but you can actually see 4 below tissue types affected
body eyes muscles brain Immune system sex tissues
a stupid idea I threw out there was maybe this was why there were originally 4 Yamanaka factors ,,but you don’t need all 4 to reprogram cells so I thought it was a stupid idea.. then you will later see that OCT4 is associated with the WRN protein!!! Holy smokes ! I
KLF-4 SOX-2 C-MYC OCT-4
| Asexual Somatic Aging System seen in men at higher rate. | Sexual/reproduction Aging System | ||
| Progeria
Defective Lamin A protein-truncated |
Werner’s Syndrome. DNA helicase WRN protein – truncated | ||
| Original to progeria alone | |||
| Coxa Valga & necrosis of head of femur (dev. defect?) | |||
| Dysplastic osteoporosis (growing bones)
(dev. defect?)
|
|||
| Symptoms of progeria-asexual somatic aging co-opted by WS-sexual aging system | Symptoms of WS-Sexual Aging System co-opted from progeria-asexual somatic aging system | ||
| Atherosclerosis | Atherosclerosis | ||
| Hypertension | Hypertension | ||
| Gray Hair | Gray Hair | ||
| Alopecia | Alopecia | ||
| Calcification of Heart Valves | Calc. of Heart Valves | ||
| Laryngeal Atrophy | Laryngeal Atrophy-WS | ||
| Loss of subcutaneous tissue | Loss of subcut. tissue | ||
| Hypermelanosis of Skin | Ignore these 2 columns below for now | Ignore these 2 columns below for now | Hypermelanosis of Skin |
| Hypogonadism | Hypogonadism -AT, XP | Hypogonadism – |
| Symptoms of Mitochondrial Diseases | Symptoms of AT, XP, CS, | Symptoms of Shared w/AT, XP, CS, & Mitochondrial Diseases | |
| Muscle Wasting-MM, N | Muscle Wasting-AT | Muscle Wasting- | |
| Neuronal Degeneration/Brain Atrophy-CP, ME, MR, K | Neuronal Degeneration/Brain Atrophy -AT, XP | Neuronal Degeneration, Brain Atrophy | |
| Basal Ganglion Calcification – D, LS | Basal Ganglion Calcification – CS | Basal Ganglion Calcification | |
| Cataracts-K | Cataracts-CS | Cataracts | |
| Diabetes-K | Diabetes-AT | Diabetes | |
| Mitochondrial-dysfunction causes Dementia/Alzheimer’s Disease-??
(but not in these acclrtd mitochondrial disease’s) |
Dementia/Alzheimer’s Disease-XP | Dementia/Alzheimer’s Disease | |
| Poor Healing -XP | Poor Healing | ||
| Skin Ulcers -XP | Skin Ulcers | ||
| Thymic Atrophy-AT | Thymic Atrophy | ||
| Scaly Skin-XP | Scaly Skin- | ||
| Somatic Cancers-XP,AT | Somatic Cancers- | ||
| Lipofuscin Accumulation-CS,XP | Lipofuscin Accumulation | ||
| Arthritis-AT | Arthritis | ||
| Peripheral Osteoporosis-CS | Peripheral Osteoporosis (growth plate closure) | ||
| Symptoms unique to Werner’syndrome | |||
| Menopause | |||
| Breast, Uterine, and Ovarian atrophy and cancer | |||
| Prostate atrophy-WS, hyperplasia-WS, and cancer | |||
| Depression? |
-Notice that the aging symptoms of progeria tend to affect men more while women seem to be affected more by the aging symptoms of Werner’s Syndrome
Progeria patients look like very old men WS-the little old lady disease
– HERE’S A HUGE CLUE! Werner’s Syndrome has all the aging symptoms of progeria (and a whole lot more)!
So, the question is – Did Werner’s Syndrome “invent” these progeria aging symptoms or did it somehow co-opt them?
I approach the answer from a co-opting perspective. I like to assume that evolution repurposes what already exists in new ways rather than create things from scratch.
How might Werner’s Syndrome co-opt progeria?
First, a little background on progeria
-Progeria is caused by a truncated Lamin A protein.
-Lamin A proteins are found in the nuclear envelope of differentiated cells and give the nucleus stiffness.
-THERE ARE NO Lamin A proteins in the nuclear envelope of many pluripotent embryonic stem cells!
–Pluripotent embryonic stem (ES) cells generated from preimplantation embryos maintain unlimited self-renewal and undifferentiated states, yet do not express Lamin A in the nuclear envelope (Bru et al., 2008; Butler et al., 2009; Constantinescu et al., 2006).
This Points to another function of Lamin A proteins- they maintain a large amount of cellular differentiation – how? This is not well researched but I believe and expect that it will be shown that
-Lamin A proteins when functioning properly hold transcription factors in place on the DNA.
-I predict you need BOTH properly functioning Lamin A proteins AND the right transcription factors in place to prevent loss of differentiation = (somatic, asexual/progeria aging) of the cell.
-The inner nuclear envelope as a transcription factor resting place . EMBO Rep. 2007 Oct; 8(10): 914–919.
Several transcriptional regulators, operating in different signal-transduction pathways, have been found to interact physically with components of the inner nuclear membrane. In general, this association seems to restrict access to their target genes and limit their transactivation or transrepression abilities. The mechanisms of inner nuclear membrane association are diverse, and include regulated associations with the nuclear lamina and integral membrane proteins. Together, these findings indicate that the inside of the nuclear envelope functions as a resting place for transcription factors and suggest a more direct role for the nuclear envelope in gene regulation than previously anticipated.
!!!!!What this means! Is that the type of aging seen in progeria can be caused by truncated Lamin A proteins, OR a shortage or absence of transcription factors (produced by Horvath’s 36 anti-aging genes?). (I believe either/or will lead to the same result)!!!!!
-What does this also imply? That Lamin A proteins functions in a way very similar to being like an extra layer of histones and chromatin condensation –keeping DNA sequestered, and keeping transcription factors in their proper places
–Progeria is a disease of loss of differentiation of the cells. In fact, the nuclear envelope of a progeria-affected cell has almost no shape or stiffness, just like, an undifferentiated embryonic stem cell!
Normal Nucleus Progeria Nucleus
-There are also little to no Lamin A proteins in the nuclear envelope of many lines of undifferentiated cancer cells:
-Embryonic carcinoma cells generally express little or no A-type Lamins. Lastly, certain adult cell types that are not fully differentiated also express little or no A-type Lamins. Consistent with these findings, Lamin A/C is not essential for cell growth and survival.
In my 1998 paper I speculated that cancer cells were simply normal cells that lost some sort of control which allowed them to ATTEMPT to revert back to the original ancient ancestor that all life forms evolved from- single cell, immortal, organisms.
I proposed this because, like the cancer cell, the single cell version of a human = the embryonic stem cell is immortal as well.
Both embryonic stem cells AND cancer cells use a form of anerobic energy generation (glycolysis-w/no oxygen required) EVEN WHEN OXYGEN IS PRESENT. (Known as the Warburg effect and once leading to the idea that lack of oxygen was the cause of cancer).
To ME THIS IS REALLY FASCINATING-WHY? I SEE IT AS OUR SHOWING US THAT OUR SINGLE CELL ANCESTORS CAN BE SEEN IN THE FIRST STEP OF HUMAN DEVELOPMENT -AT THE SINGLE CELL STAGE WHERE, LIKE OUR FIRST ANCESTOR, – OUR SINGLE CELL SELVES ARE IMMORTAL AND STILL RETAIN ENERGY METABOLISM TRAITS THAT EVOLVED BEFORE THE WORLD HAD AN OXYGEN ATMOSPHERE!
Alright So how does WS co-opt progeria? I have a good idea for this….
One fact I learned that is KEY-The Lamin A truncation seen in progeria is also seen in normal human aging at older ages.!
Lamin A-Dependent Nuclear Defects in Human Aging
SCIENCE 19 May 2006 Vol 312, Issue 5776 pp. 1059-1063
Mutations in the nuclear structural protein Lamin A cause the premature aging syndrome Hutchinson-Gilford progeria (HGPS). Whether Lamin A plays any role in normal aging is unknown. We show that the same molecular mechanism responsible for HGPS is active in healthy cells. Cell nuclei from old individuals acquire defects similar to those of HGPS patient cells, including changes in histone modifications and increased DNA damage. Age-related nuclear defects are caused by sporadic use, in healthy individuals, of the same cryptic splice site in Lamin A whose constitutive activation causes HGPS. Inhibition of this splice site reverses the nuclear defects associated with aging. These observations implicate Lamin A in NORMAL physiological aging.
Another interesting fact I learned is that human cells also use truncated Lamin A as a way to complete the development of some young developing tissues!!!
So, from an evolutionary point of view
the Lamin A truncation is no accident!
In: Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology [Internet]. Tokyo: Springer; 2016. Chapter 34.
2016 Jun 25.
See excerpt at end in extra material section
Knowing that Lamin A truncation is a normal part of aging I wondered how does it happen in older individuals- I figured it happened the same way it occurs in kids with progeria- the RNA transcript is truncated to a short version of normal Lamin A protein.
THIS LED TO A PREDICTION- Which I shared with the Conboy’s when they were looking for what substance is found in older blood that makes it toxic to younger individuals. In addition to the hormones LH and FSH and hCG.
-I suggested they look for some sort of RNA-ase that binds to and cuts the Lamin A RNA transcript to make them shorter to see how old blood might cause aging.
So, this is where Horvath’s study comes into play of the 12 genes that get turned on with aging; the one that was most widely expressed in various tissues was the LARP1 protein gene.
AND GUESS WHAT? LARP1 is unusual in that it has a section that binds to RNA!
I believe that it is through the LARP1 gene that Werner’s has coopted the progeria/ somatic, asexual aging system.
I found a telling review of LARP1 functions-it sure looks like it could be up to no good!
The role of LARP1 in translation and beyond
Wiley Interdisciplinary reviews. RNA, 17 Apr 2015, 6(4):399-417
Abstract (Truncated) We review here evidence suggesting that LARP1 are key versatile messenger RNA (mRNA)-binding proteins involved in regulating important biological processes LARP1 proteins perform many essential tasks likely by binding to key mRNAs and regulating their stability and/or translation. In human, the impact of LARP1 over cell division and proliferation is potentially under the control of the TORC1 complex. TOR-dependent LARP1 phosphorylation could specifically enhance the translation of TOP mRNAs providing a way to promote translation, growth, and proliferation. Consequently, LARP1 is found to be significantly upregulated in many malignant cell types. In plants, LARP1 was found to act as a cofactor of the heat-induced mRNA degradation process, an essential acclimation strategy leading to the degradation of more than 4500 mRNAs coding for growth and development housekeeping functions. LARP1 proteins are therefore emerging as critical ancient mRNA-binding factors that evolved common as well as specific targets and regulatory functions in all eukaryotic lineages.
Heat stressed plant
. -In summary, at this point it appears that the expression of LARP1 protein with aging in mammals allows the dominant active sexual (Werner’s) aging system to coopt the previously evolved asexual (progeria) somatic aging system most likely by truncating Lamin A RNA transcript in a manner similar to that seen in progeria.
The NEXT IMPORTANT POINT- KIDS WITH PROGERIA ALMOST NEVER GET CANCER!
I studied this question high and low for a long time and in 1998 I could not find any cases of kids with progeria with cancer. There was one case of benign prostate hypertrophy with a suspicion of some cancer but that was it! And I had to go way back in time to the basement journals to find this one case. IN 1998 there were no cases posted to Pub Med. However, after a new search, 24 years later, one case, an osteosarcoma, popped up but was dated 1978. So, after the new search of “progeria and cancer” in Pub Med (451 of them) there were two cases of bone cancer (osteosarcoma) 1978, and 2007, and one case of squamous cell cancer appearing on one progeria patient’s tongue in 2021-the third case of cancer ever reported in progeria kids!
Osteosarcoma in a patient with Hutchinson-Gilford progeria J Med Genet . 1978 Dec;15(6):481-4.
A 13-year-old female with Hutchinson-Gilford progeria, who developed an osteosarcoma of the right chest wall, is reported. This is the first reported association of a malignant neoplasm with this syndrome.
Oral Oncol. 2021 Feb;113:.Management of a rare case of squamous cell carcinoma of the tongue in a patient affected by progeria
-I have always suspected that progeria symptoms are caused mainly in a passive way and primarily through necrosis of tissues rather than through the active process of apoptosis. Atherosclerosis/Arteriosclerosis are major symptoms of progeria and lead to most deaths from the disease. Atherosclerosis generates many dying cells and they die mainly through necrosis.
-In my 1998 paper, after studying the process of apoptosis, I speculated that many if not most cancers arise from a failure of apoptosis. Apoptosis takes a lot of energy, and that apoptosis is a modified form of mitosis. In apoptosis the DNA is demethylated which removes methyl groups that protect restriction/cleavage sites, and then restriction enzymes are released that chop up the DNA. I theorized that if for some reason these little knives don’t come out and chop up the DNA, apoptosis fails and the cell may continue with the process of apoptosis/mitosis- becoming a cancer cell. IN a 2014 article, “What is apoptosis?”, cell biologist Michael Overholtzer explains that this view of apoptosis is basically correct.(see article in supplementary materials at end… To summarize-
-I believe that cancer is mostly a result of a failure of apoptosis (the active form of killing cells) and because progeria cell death is primarily caused by necrosis (the passive form of cell death) – this seems to be the explanation for why progeria patients almost never get cancer.
Liver cells blebbing
and undergoing apoptosis
How to prevent or reverse the aging symptoms seen in the progeria/asexual/somatic aging system?
We simply need to reverse what we think is causing this aging. Mainly two things- the truncation of the Lamin A protein and a shortage/deficient of the transcription factors made by Horvath’s 36 anti-aging genes. There are quite a few different ways to attack the problem. At this point we could
-Provide the aging organism with a cocktail of wild type Lamin A proteins and the 36 transcription factors produced by Horvath’s 36 anti-aging genes.
-We could try to prevent the truncation of Lamin A RNA transcripts by providing the aging organism with LARP1 anti sense RNA’s
-Or a Simple approach: We could try to shut down the LARP1 gene by reactivating the methyltransferases that stop working with aging. (This approach should also deactivate the other 11 Horvath pro-aging genes-more on this later) I believe this could be done with hormonal and NAD+ therapy (hormones to be discussed later in relation to the 12 Horvath pro-aging genes). Primarily by supplementing with high dose melatonin which declines by 90% or more with aging. Melatonin also suppresses the production of LH and FSH -the pro-aging hormones that skyrocket after age 50. (Other declining hormones that should be supplemented include DHEA, pregnenolone, and maybe progesterone) And with NAD+ AND
-Supplementing with high dose Alpha Keto glutarate and Vitamin C (vitamin C makes AKG work better with TET enzymes) This should reactivate the TET enzymes which in turn will cause the 36 Horvath anti-aging genes to become demethylated and get dialed back up.
Scientists once tried to correct progeria with wild type Lamin A and they failed, but they did correct it with CRISPR-like technology by fixing the splice site mutation-
Nat Med. 2005 Apr;11(4):440-5.
Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome
(truncated) Introduction of wild-type Lamin A protein does not rescue the cellular disease symptoms. The mutant LMNA mRNA and Lamin A protein can be efficiently eliminated by correction of the aberrant splicing event using a modified oligonucleotide targeted to the activated cryptic splice site.
-Rapamycin also seems to hold promise for treating the progeria aging system
Progeria, rapamycin and normal aging: recent breakthrough Aging (Albany NY). 2011 Jul;3(7):685-91. A recent discovery that rapamycin suppresses a pro-senescent phenotype in progeric cells not only suggests a non-toxic therapy for progeria but also implies its similarity with normal aging.
INTERMISSION
Next Topic– active sex-related/Werner’s Syndrome aging system
What causes Werner’s Syndrome? The improper (short) truncation of the WRN protein which when found in groups of up to 6 subunits has DNA helicase activity
Single sub units have no helicase activity but are involved in heterochromatin genetic transcription and gene silencing.
Above is a 6 subunit WRN DNA helicase
-Let us assume that Werner’s Syndrome represents an acceleration of normal aging seen in humans.
There is evidence that this is true:
For example:
–Aging Is Accompanied by a Progressive Decrease of Expression of the WRN Gene in Human Blood Mononuclear Cells the Journals of Gerontology: Series A, Volume 66A, Issue 1, January 2011, Pages 19–25, (truncated) The mean level of the WRN messenger RNA was significantly lower in long-living individuals than in young and middle-aged controls (p < .001 and p = .025, respectively). We suggest that age-related decrease of the WRN expression might contribute to human immunosenescence.
-In the study below, they found that cells lacking WRN protein leads to a GLOBAL loss of H3K9me3! So, therefore somehow WRN protein leads to the addition of H3K9me3 to DNA!
-What is H3K9me3? It is a gene suppressor that shuts down genes in heterochromatin with histone #3. When histone #3 has 3 methyl groups attached to its #9 lysine it prevents gene transcription. (Thus, the name H3K9me3). The interesting point here is that WRN is involved with keeping genes shut down, and when WRN is lacking-due to aging, genes get turned on.
-How does WRN keep H3K9me3 levels high in the chromatin? WRN associates with and activates the protein SUV39H1-this protein is a histone lysine methyltransferase that trimethylates lysine 9 of histone H3, which results in transcriptional gene silencing.
-Now this should be very interesting to all – Suv39h1 expression is repressed by OCT4 protein through the induction of an antisense long non-coding RNA AND WRN is involved in de novo DNA methylation of the promoter of the Oct4 gene, WRN localizes to the Oct4 promoter. It Looks like there is a war going on between WRN and OCT4
–WRN promotes the differentiation of cells through maintaining H3K9me3 silencing of genes in heterochromatin while OCT4 reverses cellular differentiation by suppressing H3K9me3 which leads to the expression of a large number of genes in the heterochromatin. It is this last point that gives me hope that we can soon unify Steve’s results, this aging theory, and Yamanaka factor rejuvenation logic into one cohesive whole.
Ok here is the abstract referenced above…
Science. 2015 Jun 5;348(6239):1160-3.
Werner syndrome (WS) is a premature aging disorder caused by WRN protein deficiency. Here, we report on the generation of a human WS model in human embryonic stem cells (ESCs). Differentiation of WRN-null ESCs to mesenchymal stem cells (MSCs) recapitulates features of premature cellular aging, a global loss of H3K9me3, and changes in heterochromatin architecture. We show that WRN associates with heterochromatin proteins SUV39H1 and HP1α and nuclear lamina-heterochromatin anchoring protein LAP2β. Targeted knock-in of catalytically inactive SUV39H1 in wild-type MSCs recapitulates accelerated cellular senescence, resembling WRN-deficient MSCs. Moreover, decrease in WRN and heterochromatin marks are detected in MSCs from older individuals. Our observations uncover a role for WRN in maintaining heterochromatin stability and highlight heterochromatin disorganization as a potential determinant of human aging.
Okay so now let’s try to figure out how Horvath’s 12 pro-aging genes, normal aging that is accelerated in Werner’s Syndrome, and the hormone changes that occur with aging are all related to the active sex-related/Werner’s Syndrome aging system.
-A big clue comes from dementia/Alzheimer’s disease which is a symptom of Werner’s Syndrome, which is also seen in “a set” of Horvath’s 12 pro-aging genes, and is found to be associated with and driven by the dramatic increases in Luteinizing Hormone seen in humans after age 50.
-See the study below and notice the name Richard Bowen-I was able to prove with 100% certainty that Bowen and others STOLE the idea of suppressing LH to treat Alzheimer’s from my 1998 paper. Bowen was given the idea that LH drives Alzheimer’s from my 1998 paper ultimately from the editor of the publishing journal David Horrobin through his associate the disgraced Dr Goran Jamal -I’ll tell you the story some day if you are interested!
Luteinizing hormone, a reproductive regulator that modulates the processing of amyloid-beta precursor protein and amyloid-beta deposition J Biol Chem
2004 May 7;279(19):20539-45.Richard L Bowen 1, Giuseppe Verdile, Tianbing Liu, Albert F Parlow, George Perry, Mark A Smith, Ralph N Martins, Craig S Atwood
Hormonal changes associated with the dysregulation of the hypothalamic-pituitary-gonadal (HPG) axis following menopause/andropause have been implicated in the pathogenesis of Alzheimer’s disease (AD). Experimental support for this has come from studies demonstrating an increase in amyloid-beta (Abeta) deposition following ovariectomy/castration. Because sex steroids and gonadotropins are both part of the HPG feedback loop, any loss in sex steroids results in a proportionate increase in gonadotropins. (truncated) These results suggest the marked increases in serum LH following menopause/andropause as a physiologically relevant signal that could promote Abeta secretion and deposition in the aging brain. Suppression of the age-related increase in serum gonadotropins using anti-gonadotropin agents may represent a novel therapeutic strategy for AD.
-Okay so we see that a disease that is seen in Werner’s syndrome dementia/Alzheimer’s is also driven by increases in the reproductive/development (free radical) hormone LH and is also seen in one of the 12 genes identified by Horvath that get activated with aging.
-Another gene that gets demethylated and activated in the 12 Horvath pro-aging genes is involved with control of circadian rhythms. Guess what else is involved with the control of circadian rhythms? The super-antioxidant hormone -Melatonin
Melatonin and circadian rhythm Rev Neurol (Paris)
2001 Nov;157(11 Pt 2):S121-5.
Melatonin is an indole hormone that is produced by the pineal gland, mainly at night, with a peak around 3.00 a.m. under normal environmental conditions. This endogenic secretion cycle is generated by the suprachiasmatic nuclei in response to the day/night alternation. Light either suppresses or entrains melatonin production according to the time of light exposure. Melatonin can be viewed as the “hand” of the internal clock and is regulated via the central nervous and sympathetic systems. Melatonin synchronizes biological cycles, particularly the temperature and sleep/wake cycles. Exogenous melatonin can influence the endogenous secretion of melatonin according to a phase response curve, an effect that provides a rationale for the use of melatonin to treat disorders of biological rhythms (rapid time-zone change syndrome, delayed sleep phase syndrome, desynchronization in blind subjects or shift workers, insomnia in the elderly).
-Also, melatonin has been found to prevent the progression of Alzheimer’s disease and can reverse menopause in women recently undergoing it! Melatonin suppresses LH and FSH and hCG and can be used as birth control in doses of 75 mg/night.
Other ANTIOXIDANT hormones that decline with age
Both sexes shown
Progesterone in women
In men the decline starts around age 60
-We also suggested that NAD+ also is active in the active sex-related/Werner’s Syndrome aging system well looky here (see below)
Nat Commun. 2019; 10: 5284
NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome
Metabolic dysfunction is a primary feature of Werner syndrome (WS), a human premature aging disease caused by mutations in the gene encoding the Werner (WRN) DNA helicase. WS patients exhibit severe metabolic phenotypes, but the underlying mechanisms are not understood, and whether the metabolic deficit can be targeted for therapeutic intervention has not been determined. Here we report impaired mitophagy and depletion of NAD+, a fundamental ubiquitous molecule, in WS patient samples and WS invertebrate models. WRN regulates transcription of a key NAD+ biosynthetic enzyme nicotinamide nucleotide adenylyltransferase 1 (NMNAT1). NAD+ repletion restores NAD+ metabolic profiles and improves mitochondrial quality through DCT-1 and ULK-1-dependent mitophagy. At the organismal level, NAD+ repletion remarkably extends lifespan and delays accelerated aging, including stem cell dysfunction, in Caenorhabditis elegans and Drosophila melanogaster models of WS. Our findings suggest that accelerated aging in WS is mediated by impaired mitochondrial function and mitophagy, and that bolstering cellular NAD+ levels counteract WS phenotypes.
I am running out of time so I will skip a few points I wanted to make here in the discussion points. The points were about how the rapid aging segmental diseases of Cockayne Syndrome, Xeroderma Pigmentosum, Ataxia Telangiectasia, and the many mitochondrial diseases that cause rapid segmental aging (see my prior exhibit of the symptoms of aging vs various diseases table) and just throw some big ones at you for now.
-The asexual/somatic /Progeria aging system was previously described as a passive form of somatic aging driven primarily through loss of differentiation leading to necrosis of tissues.
-The active sex-related/Werner’s Syndrome aging system is an active aging system designed to either shut down an individual’s reproduction (i.e., menopause and/or kill the individual. Its primary goal is cessation of reproduction by an all-out attack on reproductive tissues liked the uterus, breasts, prostate etc. It does this by triggering the ACTIVE form of cell death: APOTOSIS.
-I believe that this assault on the reproductive tissues (mostly by LH and also FSH and hCG) also has the side effects of causing apoptosis to occur in other tissues like the brain, skin, nerves, bones, everything!
-It turns out after I wrote my 1998 paper that researchers were surprised when they found LH receptors in every tissue in the body- I wasn’t.
-So, the triggering of apoptosis and atrophy in all these tissues leads to what one could call “the little old lady syndrome”. When apoptosis malfunctions it leads to the many cancers you see in old age and Werner’s Syndrome and the segmental non-progeria diseases
-Finally, Horvath noticed that in his study he found genes that were affected by aging included genes for
-Age at menarche
-Gestation time
-Body size
-Longevity /Maximum Lifespan
This makes sense from my point of view. Given that aging is programmed you have to ask why would evolution want to kill us? The answer I believe is to stop us from reproducing so we do not contribute too much too the gene pool!
If this motive of evolution is correct then it would explain something we see in nature amongst almost all animals.
All these traits are linked>>>>
Body size, Litter size, Gestation Time, Age at sexual maturity, and maximum lifespan.
So, if you pull on maximum lifespan and increase it in a species it tends to drag along all the other factors in most species:
Body Size, litter size, gestation time, sexual maturity age, max lifespan
Body Size increases, litter size decreases, gestation time increases, time to sexual maturity increases
What do these things have in common? A reduction in the maximum number of offspring one can have in a lifetime!
2 more points
- Prediction: A very good aging clock will be found by looking at H3K9me3’s>>> Histone 3’s with trimethylated lysine 9’s
Now this is kind of a problem to explain at the moment ….
- The fact that a FSH and LH surge triggers the normal development (see below) that triggers accelerated aging/development in progeria suggests that the asexual/somatic passive /progeria aging system is under some sort of hormonal control….I have always suspected FSH for this role as I looked high and low and could find no cancers associated with FSH while I found huge numbers of cancers associated with LH
I have run out time for the discussion points for now….
Please enjoy the following
Supplementary Information at your leisure
Supplementary Materials
Aging Is Accompanied by a Progressive Decrease of Expression of the WRN Gene in Human Blood Mononuclear Cells
The Journals of Gerontology: Series A, Volume 66A, Issue 1, January 2011, Pages 19–25,
The WRN gene encodes DNA helicase participating in genome maintenance. We looked for associations of natural aging with expression and methylation of this gene in blood mononuclear cells and with its common polymorphisms. Analyses were performed in ethnically homogenous Polish Caucasians. The mean level of the WRN messenger RNA was significantly lower in long-living individuals than in young and middle-aged controls (p < .001 and p = .025, respectively). Analysis of the 361 bp WRN promoter CpG island showed that aging might be accompanied by a slight increase of its methylation status; however, it seems to be biologically insignificant. Finally, analysis of the WRN R834C, L1074F, and C1367R polymorphisms showed that the frequencies of the L1074F and C1367R polymorphisms were similar in all age groups tested, whereas the R834C polymorphism was absent from Polish Caucasians. We suggest that age-related decrease of the WRN expression but not its common genetic variants might contribute to human immunosenescence.
Aging Cell
A role for the Werner syndrome protein in epigenetic inactivation of the pluripotency factor Oct4
Aging Cell . 2010 Aug;9(4):580-91.
Werner syndrome (WS) is an autosomal recessive disorder, the hallmarks of which are premature aging and early onset of neoplastic diseases (Orren, 2006; Bohr, 2008). The gene, whose mutation underlies the WS phenotype, is called WRN. The protein encoded by the WRN gene, WRNp, has DNA helicase activity (Gray et al., 1997; Orren, 2006; Bohr, 2008; Opresko, 2008). Extensive evidence suggests that WRNp plays a role in DNA replication and DNA repair (Chen et al., 2003; Hickson, 2003; Orren, 2006; Turaga et al., 2007; Bohr, 2008). However, WRNp function is not yet fully understood. In this study, we show that WRNp is involved in de novo DNA methylation of the promoter of the Oct4 gene, which encodes a crucial stem cell transcription factor. We demonstrate that WRNp localizes to the Oct4 promoter during retinoic acid-induced differentiation of human pluripotent cells and associates with the de novo methyltransferase Dnmt3b in the chromatin of differentiating pluripotent cells. Depletion of WRNp does not affect demethylation of lysine 4 of the histone H3 at the Oct4 promoter, nor methylation of lysine 9 of H3, but it blocks the recruitment of Dnmt3b to the promoter and results in the reduced methylation of CpG sites within the Oct4 promoter. The lack of DNA methylation was associated with continued, albeit greatly reduced, Oct4 expression in WRN-deficient, retinoic acid-treated cells, which resulted in attenuated differentiation. The presented results reveal a novel function of WRNp and demonstrate that WRNp controls a key step in pluripotent stem cell differentiation.
Nat Commun. 2019; 10: 5284
NAD+ augmentation restores mitophagy and limits accelerated aging in Werner syndrome
Metabolic dysfunction is a primary feature of Werner syndrome (WS), a human premature aging disease caused by mutations in the gene encoding the Werner (WRN) DNA helicase. WS patients exhibit severe metabolic phenotypes, but the underlying mechanisms are not understood, and whether the metabolic deficit can be targeted for therapeutic intervention has not been determined. Here we report impaired mitophagy and depletion of NAD+, a fundamental ubiquitous molecule, in WS patient samples and WS invertebrate models. WRN regulates transcription of a key NAD+ biosynthetic enzyme nicotinamide nucleotide adenylyltransferase 1 (NMNAT1). NAD+ repletion restores NAD+ metabolic profiles and improves mitochondrial quality through DCT-1 and ULK-1-dependent mitophagy. At the organismal level, NAD+ repletion remarkably extends lifespan and delays accelerated aging, including stem cell dysfunction, in Caenorhabditis elegans and Drosophila melanogaster models of WS. Our findings suggest that accelerated aging in WS is mediated by impaired mitochondrial function and mitophagy, and that bolstering cellular NAD+ levels counteracts WS phenotypes.
Front. Cell Dev. Biol., 01 June 2022 |
ATM Modulates Nuclear Mechanics
by Regulating Lamin A Levels
Ataxia-telangiectasia mutated (ATM) is one of the three main apical kinases at the crux of DNA damage response
and repair in mammalian cells.
Nuclear deformation characterizes
Werner syndrome cellsJanuary 2006
Basic Res Cardiol
. 2011 Sep;106(5):749-60.
Necrotic cell death in atherosclerosis
Necrosis is a type of cell death characterized by a gain in cell volume, swelling of organelles, rupture of the plasma membrane and subsequent loss of intracellular contents. For a long time, the process has been considered as a merely accidental and uncontrolled form of cell death, but accumulating evidence suggests that it can also occur in a regulated fashion. Morphological studies using transmission electron microscopy indicate that the vast majority of dying cells in advanced human atherosclerotic plaques undergo necrosis. Various stimuli in the plaque including high levels of oxidative stress, depletion of cellular ATP, impaired clearance of apoptotic cells and increased intracellular calcium may cause necrotic death. Although the role of necrosis in atherosclerosis remains ill-defined, a growing body of evidence suggests that necrotic death stimulates atherogenesis through induction of inflammation and enlargement of the necrotic core. In addition, necrosis contributes to plaque instability by releasing tissue factor, matrix degrading proteases and pro-angiogenic compounds. Therapeutic agents against necrosis are limited, but efforts have recently been made to inhibit the necrotic pathway or its pro-inflammatory effects.
What Is Apoptosis?
By Julie GrishamFriday, May 16, 2014
Liver cells blebbing and undergoing apoptosis
Summary
Cell biologist Michael Overholtzer explains apoptosis, a form of programmed cell death that can lead to cancer when it doesn’t function properly.
The death of one tiny cell might seem like a simple thing. But the process is much more complicated than you would expect.
There are actually several types of cell death, each with its own unique characteristics and processes. One of the most well-studied is called apoptosis.
“Apoptosis is defined by a set of physical, often visible, features that are associated with the demise of an individual cell,” says Memorial Sloan Kettering cell biologist Michael Overholtzer. “It’s probably one of the most common forms of cell death during the development of an organism. It also plays an important role in cancer.”
One purpose of apoptosis is to eliminate cells that contain potentially dangerous mutations. If a cell’s apoptosis function is not working properly, the cell can grow and divide uncontrollably and ultimately create a tumor.
Visible Changes, Molecular Changes
So what do you see if you look at an apoptotic cell under a microscope? “The nucleus looks shrunken or condensed and fragmented into pieces, whereas a normal nucleus is one round oval,” Dr. Overholtzer explains.
“The other obvious feature is that the cells themselves would shrink and begin to bleb,” he adds. “Blebbing means that the cell’s membrane changes, and there are bulging protrusions from the surface of the cell.”
An enzyme called caspase starts the chain reaction of changes that lead to a cell’s death by apoptosis. “Caspase is essentially like molecular scissors,” Dr. Overholtzer says. “In a normal, happy cell, it’s inactive, but once a cell is either put under stress or developmentally programmed to commit suicide, the scissors are activated and start to cut up certain proteins inside the cell, beginning the apoptosis process.”
In cancer cells, however, the scissors may not get the signal to start cutting. “The sensor that recognizes cell damage may not work, and the signal is never sent,” he says. “There are currently strategies under way to develop drugs that would help reactivate the sensor and therefore activate apoptosis.”
One of those sensors is a well-studied protein called p53. When it functions normally, it suppresses the formation of cancer through apoptosis. Mutations in the p53 gene are found in about half of all cancers.
In: Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology [Internet]. Tokyo: Springer; 2016. Chapter 34.
2016 Jun 25.
Excerpt
The ductus arteriosus (DA) is a fetal vessel bypassing the still nonfunctional lungs. Closure of the DA at birth is essential for the transition from a fetal to a neonatal circulation. This closing process begins with a physiological contraction followed by definitive anatomical closure. The latter process starts already before birth by development of intimal thickening followed after birth by degeneration of the inner media, including cytolytic necrosis and apoptosis. The histological changes during normal DA closure resemble the features seen in the premature ageing vessels in children with the Hutchinson progeria syndrome. The latter syndrome is caused by a mutation in the Lamin A/C gene resulting in accumulation of the progerin splice variant. We studied human DA biopsies from the fetal to the neonatal period to investigate whether Lamin A/C and progerin might be involved in the DA closure process. The results show an increase in the intima and inner media of progerin in the normal neonatal DA, while expression of Lamin A/C is diminished. In the non-closing aorta, the fetal DA and the PDA, no or hardly any progerin expression was found. We postulate that the Lamin A/C to progerin balance is important during normal anatomical closure of the DA presenting a unique case of physiological premature vascular ageing.
Int J Biochem Cell Biol
. 2005 May;37(5):947-60. Epub 2004 Dec 15.
Genetic alterations in accelerated ageing syndromes. Do they play a role in natural ageing?
The molecular mechanisms leading to human senescence are still not known mostly because of the complexity of the process. Different research approaches are used to study ageing including studies of monogenic segmental progeroid syndromes. None of the known progerias represents true precocious ageing. Some of them, including Werner (WS), Bloom (BS), and Rothmund-Thomson syndromes (RTS) as well as combined xeroderma pigmentosa-Cockayne syndrome (XP-CS) are characterised by features resembling precocious ageing and the increased risk of malignant disease. Such phenotypes result from the mutations of the genes encoding proteins involved in the maintenance of genomic integrity, in most cases DNA helicases. Defective functioning of these proteins affects DNA repair, recombination, replication and transcription. Other segmental progeroid syndromes, such as Hutchinson-Gilford progeria (HGPS) and Cockayne syndrome are not associated with an increased risk of cancer. In this paper we present the clinical and molecular features of selected progeroid syndromes and describe the potential implications of these data for studies of ageing and cancer development.
WHY NO cancer in Cockayne Syndrome? See below CSB more like progeria loss of differentiation via loss of H3K9me3
Nucleic Acids Res. 2019 Sep 19; 47(16): 8548–8562.
Cockayne syndrome group B deficiency reduces H3K9me3 chromatin remodeler SETDB1 and exacerbates cellular aging
Cockayne syndrome is an accelerated aging disorder, caused by mutations in the CSA or CSB genes. In CSB-deficient cells, poly (ADP ribose) polymerase (PARP) is persistently activated by unrepaired DNA damage and consumes and depletes cellular nicotinamide adenine dinucleotide, which leads to mitochondrial dysfunction. Here, the distribution of poly (ADP ribose) (PAR) was determined in CSB-deficient cells using ADPr-ChAP (ADP ribose-chromatin affinity purification), and the results show striking enrichment of PAR at transcription start sites, depletion of heterochromatin and downregulation of H3K9me3-specific methyltransferases SUV39H1 and SETDB1. Induced-expression of SETDB1 in CSB-deficient cells downregulated PAR and normalized mitochondrial function. The results suggest that defects in CSB are strongly associated with loss of heterochromatin, downregulation of SETDB1, increased PAR in highly-transcribed regions, and mitochondrial dysfunction.
Aberrant DNA methylation profiles in the premature aging disorders Hutchinson-Gilford Progeria and Werner syndrome . 2013 Jan;8(1):28-33.
Global Transcription in Pluripotent Embryonic Stem Cells
Open Chromatin and Hypertranscription in Embryonic Stem Cells
Cell Stem Cell, Volume 2, Issue 5, 8 May 2008, Pages 408-410
The molecular mechanisms underlying pluripotency and lineage specification from embryonic stem cells (ESCs) are largely unclear. Differentiation pathways may be determined by the targeted activation of lineage-specific genes or by selective silencing of genome regions. Here we show that the ESC genome is transcriptionally globally hyperactive and undergoes large-scale silencing as cells differentiate. Normally silent repeat regions are active in ESCs, and tissue-specific genes are sporadically expressed at low levels. Whole-genome tiling arrays demonstrate widespread transcription in coding and noncoding regions in ESCs, whereas the transcriptional landscape becomes more discrete as differentiation proceeds. The transcriptional hyperactivity in ESCs is accompanied by disproportionate expression of chromatin-remodeling genes and the general transcription machinery. We propose that global transcription is a hallmark of pluripotent ESCs, contributing to their plasticity, and that lineage specification is driven by reduction of the transcribed portion of the genome.
Aging Cell
A role for the Werner syndrome protein in epigenetic inactivation of the pluripotency factor Oct4
Aging Cell . 2010 Aug;9(4):580-91.
Werner syndrome (WS) is an autosomal recessive disorder, the hallmarks of which are premature aging and early onset of neoplastic diseases (Orren, 2006; Bohr, 2008). The gene, whose mutation underlies the WS phenotype, is called WRN. The protein encoded by the WRN gene, WRNp, has DNA helicase activity (Gray et al., 1997; Orren, 2006; Bohr, 2008; Opresko, 2008). Extensive evidence suggests that WRNp plays a role in DNA replication and DNA repair (Chen et al., 2003; Hickson, 2003; Orren, 2006; Turaga et al., 2007; Bohr, 2008). However, WRNp function is not yet fully understood. In this study, we show that WRNp is involved in de novo DNA methylation of the promoter of the Oct4 gene, which encodes a crucial stem cell transcription factor. We demonstrate that WRNp localizes to the Oct4 promoter during retinoic acid-induced differentiation of human pluripotent cells and associates with the de novo methyltransferase Dnmt3b in the chromatin of differentiating pluripotent cells. Depletion of WRNp does not affect demethylation of lysine 4 of the histone H3 at the Oct4 promoter, nor methylation of lysine 9 of H3, but it blocks the recruitment of Dnmt3b to the promoter and results in the reduced methylation of CpG sites within the Oct4 promoter. The lack of DNA methylation was associated with continued, albeit greatly reduced, Oct4 expression in WRN-deficient, retinoic acid-treated cells, which resulted in attenuated differentiation. The presented results reveal a novel function of WRNp and demonstrate that WRNp controls a key step in pluripotent stem cell differentiation.
Update #35 Fantastic Confirming New Studies Just found -updated table
ATM protein stimulates Lamin A production! (An example of subsequently evolved aging systems coopting previously evolved aging systems.#3 coopts #1..)..Also the proteins defective in AT, CS, and XP are all found inside the mitochondria (another example of aging system #3 coopting aging system #2)..Also ATM (mutated ) protein causes accelerated aging in the cell, when removed form the mitochondria senescence is reversed
TET ENZYMES ONLY ACTIVE IN PRESENCE |DNA METHYLTRANSFERASES WORK IN 2
OF ANTIOXIDANTS AKG AND VITAMIN C | DIRECTIONS IN THE PRESENCE OF ANTI
<Aging from Loss of Differentiation> | <Aging from Active Atrophy of Tissues>
KLF4 AS#1 SOX2 AS#2 c-MYC AS#3 OCT-4 AS#4
(aka SRY=malesexgene) BIG TIME (& a little c-myc)
PROGERIA MITOCHONDRIAL XERODERMA PIGM WERNER'S
(+some c-myc) AGING DISEASES COCKAYNE SYNDR SYNDROME
Ataxia Telangiectasia
lots of apoptosis
|
Aging System #1 Loss of Cellular Differentiation seen in men at higher rate.
NECROSIS
|
Aging System #2 Somatic atrophy: Mitochondrial Apoptosis,
|
Aging System #3 Somatic atrophy +Immune senescence: Apoptosis, LH/hCG driven, seen in women at a higher rate APOPTOSIS
|
Aging System # 4 Sex tissue atrophy: LH, FSH, hCG
estrogen/DHT driven, seen in women at higher rate (co-opts #3, #2, (and #1)) APOPTOSIS
|
|
Progeria only.
Truncated Lamin A protein aging system #1
|
Mitochondrial Myopathy (MM), NARP (N), CPEO (CP), MELAS (ME), MERRF (MR) , KSS (K), Dystonia (D), Leigh’s Syndrome (LS) aging system #2
|
Ataxia Telangiectasia (AT), Xeroderma Pigmentosum (XP), Cockayne Syndrome (CS)..
aging system #3 cooopts #2 by entering mitochindria AT coopts #1 by stimulating Lamin A production
|
Werner’s Syndrome. (WS),
Truncated WRN protein
aging system #4
|
|
Original to #4 alone (likely defects of development)
|
|||
|
Coxa Valga & necrosis of head of femur
|
|||
|
Dysplastic osteoporosis
|
|||
|
Symptoms of #1 co-opted by #4
|
Symptoms of #4 co-opted from #1
|
||
|
Atherosclerosis
|
Atherosclerosis-WS
|
||
|
Hypertension
|
<<<<<<<<<<< Aging System #4 Co-Opts #1 (and probably #3)
|
Via LARP1 protein binding to and truncating Lamin A RNA
|
Hypertension-WS
|
|
Gray Hair
|
Gray Hair-WS
|
||
|
Alopecia
|
Alopecia-WS
|
||
|
Calcification of Heart Valves
|
Calc. of Heart Valves-WS
|
||
|
Laryngeal Atrophy
|
Laryngeal Atrophy-WS
|
||
|
Loss of subcutaneous tissue
|
Loss of subcut. tissue-WS
|
||
|
Hypermelanosis of Skin
|
Hypermelanosis of Skin-WS
|
||
|
Hypogonadism (defect of development?)
|
Hypogonadism -AT, XP
|
Hypogonadism -WS
|
|
|
Telomere Shortening
|
More research needed MELAS (ME)
|
Telomere Shortening-AT
|
Telomere Shortening-WS
|
|
Misshapen Nuclei
|
|
Hyaluronuria- excessive loss of hyaluronic acid in urine |
Misshapen Nuclei Dystonia (D) -more research needed
|
Misshapen Nuclei-AT
|
Misshapen Nuclei-WS
|
|
Hyaluronuria-excessive loss of hyaluronic acid in urine |
|
Symptoms of #2 also seen in and co-opted by #3 and #4
|
Symptoms of 3 also seen in #2 and co-opted by #4
|
Symptoms of #4 co-opted from #2 and #3
|
|
|
Muscle Wasting-MM, N
|
Muscle Wasting-AT
|
Muscle Wasting-WS
|
|
|
Neuronal Degeneration/Brain Atrophy-CP, ME, MR, K
|
|
Deafness-CPEO, MM, (ME),D
|
Neuronal Degeneration/Brain Atrophy -AT, XT
|
|
|
Deafness- XP, CS
|
Neuronal Degeneration, Brain Atrophy -WS ??
|
|
|
Basal Ganglion Calcification – D, LS
|
Basal Ganglion Calcification – CS
|
Basal Ganglion Calcification -WS
|
|
|
Cataracts-K
|
Cataracts-CS
|
Cataracts-WS
|
|
|
Diabetes-K
|
Diabetes-AT
|
Diabetes-WS
|
|
|
Mitochondrial Induced AD?
|
Alzheimer’s Disease-XP ?
|
Alzheimer’s Disease-WS ??
|
|
|
Symptoms of #3 co-opted by #4
|
Symptoms of #4 co-opted from #3
|
||
|
Poor Healing -XP
|
Poor Healing -WS
|
||
|
Skin Ulcers -XP
|
Skin Ulcers -WS
|
||
|
Thymic Atrophy-AT
|
Thymic Atrophy-WS
|
||
|
Scaly Skin-XP
|
Scaly Skin-WS
|
||
|
Somatic Cancers-XP,AT
|
Somatic Cancers- WS
|
||
|
Lipofuscin Accumulation-CS,XP
|
Lipofuscin Accumulation-WS
|
||
|
Arthritis-AT
|
Arthritis-WS
|
||
|
Peripheral Osteoporosis-CS
|
Peripheral Osteoporosis-WS
|
||
|
Symptoms unique to #4
|
|||
|
Menopause-WS
|
|||
|
Breast, Uterine, and Ovarian atrophy and cancer-WS
|
|||
|
Prostate atrophy-WS, hyperplasia-WS, and cancer-WS
|
|||
|
Depression-WS?
|
(LHFPL4 OR CELF6 OR LHFPL3 OR EVX2 OR FOXB1 OR PRDM13 OR ZIC5 OR CELF4 OR LARP1 OR DLX6-AS1 OR ZIC4 OR (OTP NOT program NOT product NOT Ottawa NOT pollutant NOT caffeine NOT treatment NOT orthopantomograms NOT operating NOT opioid NOT ovine NOT primers) OR DBX1 OR (C21orf50 OR DBP5 OR KIAA1019 OR NREBP OR BASS1) OR IRX1 OR NRN1 OR GRIK2 OR POU3F2 OR SNX1 OR NR2E1 OR TLX3 OR VSX2 OR TBX18 OR OTX1 OR TCF12 OR SALL1 OR ZIC1 OR ZIC2 OR SIX2 OR HOXA13 OR NEUROG2 OR EGR3 OR FOXD3 OR (OBI1-AS1 OR BRN3a OR C13orf7 OR RNF219 OR POU4F1) OR PHOX2B OR NEUROD1 OR PAX2 OR PAX5 OR HDAC2 OR TWIST1 OR CTCF OR NKX2 OR PRC2 OR NANOG OR BDNF OR “c-JUN”)
Must run separately:> to fix the REST and transferrin problems
(((“RE-1 silencing transcription factor” OR NRSF) AND REST) NOT “during rest” NOT “balance rest” NOT “harrow rest” NOT “lay to rest” NOT “rest questions” NOT “rest of” NOT “after rest” NOT “during rest” NOT “rest in” NOT “rest tension” NOT “with rest” NOT “at rest” NOT “rest pain” NOT “the rest” NOT “bed rest”)
(transferrin AND gene) NOT hepcidin NOT targeted NOT tested NOT test NOT vitamin D3 NOT C-reactive protein NOT urinary NOT albumin NOT anemia
Notes:
To fix the SON problem- remove SON replace with SON aliases = C21orf50 OR DBP5 OR KIAA1019 OR NREBP OR BASS1 (this has been done in the main search tool)
Fix TF problem replace TF with transferrin (this requires to run TF separately).
Fix the REST problem by running the rest search terms above separately.
interesting article and brilliant analysis!!…quite paradoxical to other schools ( even lifeextensionists)….my questionists is do u have hint on “how to reactivate the 36 genes that are shut down by methylation and how to suppress that LARP1 gene to allow us to return back to a more differentiated version of ourselves”. I’m just curious to what youthink, since your high intuitions predicted this close since 1998 without sophisticated data..
Hello there…
well I think the upstream controllers of these methylation changes will be found to be hormones….some drop dramtically after age 50 soem rise dramatically check out this
article at my website>>> https://jefftbowles.com/the-6-changes-in-lifetime-hormone-levels-that-cause-aging-and-how-to-easily-reverse-them/
How could we possible know that it is hormone changes driving aging, and not aging driving hormone changes? I do not say rebalancing hormones to a more youthful state could not be beneficial, but it seems a stretch to say they are the cause of aging. To do that we’d need to see what the signal is that causes them to go awry. It could just be a feedback mechanism that gradually fails.
Good question- and valid question..but after 35+ years of research on the question..I have found that it is the hornones that drive the aging process
It is a bit slow to see in humans..so instead why dont you take a look at the atlantic salmon..who rapidly age and die in 3 days after spawning
check out their hormone changes you will see LH shooting up 10,000 percent
also check out the Pacific Salmon who also show the rapid aging and death after spawening and only live 3 years..
you cut out their sex hormone producing glands and they live 7 years.
I understand what you are saying, but I’d still like to know WHY hormone levels change so much. In the various Salmon cases you could either argue that the massive hormone boost is required to make the arduous journey back up rivers. Or perhaps it is intentionally to kill the salmon and make way for their young. It is plausible that a similar, albeit slower signal could be occurring in humans. But what is driving it? How does the body know when to turn on the kill switch?
Regarding LH and FSH, is there a recommended range that older folks should aim for?
HI there..good points/questions. One way I look at it is rather than ask if aging is programmed..Ask if menopause appears programmed. You would agree that that is true right? Well menopause just so happens to be the evolutionary equivalent of death . Once you cannot reproduce from either menopause death or aging, evolution does not care about you and you can have no more effect on the gene pool.How is menopause triggered? By a huge drop in melatonin levels. Melatonin suppresses LH and FSH. LH and FSH are very important hormones that control women’s reproductive cycle ona monthly basis. At menopause these hormoens do not go down. they shoot sky high..! And the same thing happens in men. If a women is just eentereing menopause, melatonin can reverse the process. For how long I don’t know that would make a great study. But what I can say is taking high doses of melatonin at night (75 mg + for women and maybe 120 mg + for men -due to weight differences) will help keep LH and FSH suppressed.
This seems similar to Walter Pierpaoli’s research decades ago on melatonin as the master regulator of the hormonal aspects of programmed aging, of the master aging clock.
Hi there yeah pretty similar but this takes it a lot furhter !! HAHA
Funny , my uncle (godfather) George Solomon MD the famous psychiatrist
was good friends with Walter Pierpoli and he told me to send my
1998 paper to him…Never got any response back
he probably didn’t understand it!
Where do you find such high dosage supplements of melatonin?
The highest i can find in my area is 5 mg.
HI there it is best to buy bulk powder it is much cheaper (90%!) and just put it under your tongue at night right before bed>>
check out this blog post>>> https://jefftbowles.com/how-to-save-90percent-and-more-on-vitamins-hormones-supplements/
Jeff
Does Melatonin poser taste a bit chalky flavor?
Just confirming I got what they claimed it was
How do you measure it (eg 10mg, 20mg, 75mg, 100mg etc) given the “standard” dose in pills 3mg is tiny
Do you use a sensitive electronic scale?
HI there
yeah melatonin does not have much taste where did you get yours from….
If you bought bulk powder from peter at vitaspace.com he gives you little measuring spoons…
Jeff, how much powdered melatonin should one take in MG? I know there are no set rules here, but for a 50 year old man, what dose in mg would you suggest?
hi there
All I can say is I have tried many differnt doses over the years and I have finally settled on what I think is about 400 mg of powder under my tongue every night before bed…abut as much as I can handle wihtout puking
if you do it…
but that is going to probsably make you sleep likle crazy for up to 3 months before you get used to it and there will likely be short term sexual side effects..kibd of like propecia …
Hey if you want to participate in a mini study we can get your DNA methylation age tested now before you try it and then 3 to 4 months later….are you game?? I would do it but Ive been taking too much melatonin for too long…
heck yeah, that sounds like.great fun
im game if the study is.still. rolling.
Jeff, I may be interested in the melatonin study but may have some confounding factors. I am 67 and take bio-identical estrogen/testosterone and progesterone. No DHEA or pregnenolone. I have been taking melatonin at night but only 1.5 mg dose.
HI there great news Ill sned you an email
my theory again around microbiome and microb! I read some where that the virus and microb, bacteria should be in body all intact and they just try to curb diseased cells! I wonder if this idea help with senescence cells removal from body so take out a lot of inflammatory source from body!?
I think this might interest you (it’s about Horvath’s most recent research) , can you comment on that? Do you think we could somehow emulate this effect without blood plasma (at least somewhere in the future)?
https://longlifeandhealth.org/is-the-fountain-of-youth-found-in-blood-plasma/
HI there yeah Harold Katcher is getting a patent on some plant orthologs of yamanaka factors to give you a blast of anti aging
effects with no need for blood transfusion..It should be out soon
You might get some good effects by just reversign the hormoes changes that occurred with aging…
hgh dose melatonin which is a great antioxidant but also suppresses LH and FSH…pregnenolone dhea progesterone
I have a question. Do we need to stop Vitamin D3 supplementation if we happen to be on vacation where we spend 2-3 hours at the beach with just a bathing suit?
I vacation in Spain for 2-3 weeks in the summer and spend it at the beach almost every day. Can we over do it if we are also supplementing with D3? or can we still supplement while also spending time in the Sun.
HI there
If you are in the summer sun you don’t have to take as much D3 as your skin will make about 20,000 with 1/2 hour of sunbathing but it is good to take d3 before you get out in the sun..as many people say it prevents sunburn. Interestingly they found that Israeli lifeguards had about a 7x higher incidence of kidney stones than the normal population which means they were generating excess calcium in their blood form the d3 being made in their skin and this caused kidney stones because they weren’t taking vitamin k2!!
I have found that getting glutathione shots prevent me from getting sunburned.
Large doses of vitamin C are very protective of sunburn also.
Hey Jeff:
Are you an alien, or just a genius???
I’ve toodling around with UBI, UV cell phone diodes (with DMSO for < scattering)….to get the UV INSDIE the radial artery. ANy suggestions?
Also, did you know putting UVC onto shrooms boosts their vit D levels….600 fold? (they also generate vit D4). UV light now being used by more & more food processors…..to lengthen shelf life & disinfect. But it does much more – similar to "Sun-Drying".
thanks! HAHA sorry no idea how to get uv light inside your body ..but I have always wondered what would happen if you shined UV light inside the body beneath the skin let me know if you figure it out might need a cool endoscope doctor who will let you attach and insert a fiber optic thread that emits UV light