Abstract
What if cancer cells are more than just rogue mutations—what if they represent a startling evolutionary throwback to our single-celled ancestors? In this paper, we uncover striking parallels between embryonic stem cells (ESCs) and cancer cells, focusing on their shared reliance on glycolysis (the Warburg effect), absence or minimal expression of lamin A, and capacity for indefinite self-renewal. We further reveal how these features mirror primordial life forms that predate Earth’s oxygenation. By exploring the evolutionary sequence—mitosis first, followed by sophisticated DNA repair and apoptosis—we illuminate how metabolic insufficiency in mitochondria can stall the apoptosis program, unleashing unregulated proliferation reminiscent of ancient single-celled behaviors. Building on Thomas Seyfried’s metabolic theory of cancer, we posit that restoring robust mitochondrial function might reverse cancer cells’ atavistic shift or even trigger their long-delayed cell death. From the subtle epigenetic changes that accompany mitotic chromosome segregation to the universal vulnerability of DNA under conditions of compromised energy, our synthesis bridges molecular biology, evolutionary theory, and clinical oncology. Readers will discover compelling evidence that cancer may be, at its core, a metabolic disease—one that seizes upon ancestral cellular states to circumvent modern-day apoptotic defenses. This perspective not only reframes our understanding of cancer’s origins but also promises novel therapeutic avenues targeting mitochondrial metabolism, inspiring us to look backward in order to move medicine forward.
Abstract
Monoamine Oxidase A (MAO-A) and Monoamine Oxidase B (MAO-B) are flavin-dependent enzymes that progressively increase with age in many tissues. It is proposed that both serve as “death genes,” depleting Flavin Adenine Dinucleotide (FAD) and thereby reducing mitochondrial energy production—mirroring the known action of CD38, which depletes Nicotinamide Adenine Dinucleotide (NAD+). Although MAO-A retains certain developmental and sex-related roles, MAO-B appears to confer no clear early-life benefit and emerges as the first true, fully dedicated death gene documented. This discovery challenges classical evolutionary theories and suggests an unexpected “programming” of aging. The contrasting knockout phenotypes are detailed—dramatic aggression and neurotransmitter imbalance for MAO-A vs. subtle or minimal deficits for MAO-B. How the parallel depletion of NAD+ (by CD38) and FAD (by MAOs) undermines electron transport chain function in a near-symmetric manner is also examined. These findings open new therapeutic possibilities, including targeted inhibition of MAO-B (and MAO-A) and combined strategies preserving both NAD+ and FAD to mitigate age-related decline. This finding also calls into question a core principle of the selfish gene theory of evolution and suggests a need for a reevaluation of mainstream theory.
Abstract
In this study, we unveil a universal blueprint of aging by analyzing Horvath’s 48 pivotal epigenetic aging genes alongside their prevalence in PubMed searches for key aging-related terms. Our data reveal a two-tiered genetic architecture: a core group of epigenetic “hubs” (including HDAC2, PRC2, c‐JUN, CTCF, and NANOG) that consistently surface across multiple conditions—from progeria to mitochondrial dysfunction—and a series of niche-specific genes that exhibit striking condition-targeted spikes. These findings suggest that while a handful of master regulators orchestrate the broad symphony of cellular senescence, other genes fine-tune specific pathways, such as neurodegeneration, cancer, and hormonal dysregulation. By mapping these differential patterns, our work provides a comprehensive framework that not only deepens our understanding of the molecular drivers of aging but also spotlights promising targets for therapeutic intervention. This “genetic symphony” of senescence, with its universal chords and specialized solos, offers fresh insights into the evolutionary conservation of aging processes and paves the way for innovative strategies in aging research.
Abstract
Recent studies, including Horvath’s landmark universal epigenetic clock (August 2023) and subsequent comparative analyses, highlight four deeply conserved “plant-present” genes—HDAC2, PRC2, SNX1, and LARP1—as key regulators of aging across eukaryotes. Notably, HDAC2 and PRC2 also appear in searches relating to “lamin A”/“progeria,” suggesting that premature aging syndromes may co-opt epigenetic systems first established in plant-like ancestors. SNX1 and LARP1 would also be expected to be found associated with progeria/lamin a but likely not enough studies exist for these genes for studies to appear in this context yet. In parallel, mitochondrial-centered queries underscore genes like c-JUN and HDAC2 as top hits for mammalian mitochondrial aging, reflecting broad conservation with insect aging pathways. Here, we integrate new insights from two recent articles—“Primordial Pathways of Aging” (Feb 2025) and “The Four Horsemen of Aging” (Jan 2025)—to illustrate how these universal genes bridge plant vascular senescence and metazoan aging modules. We argue that progeria (and other accelerated aging syndromes) exploits fundamental chromatin and mitochondrial regulatory circuits with roots in our earliest eukaryotic forebears.
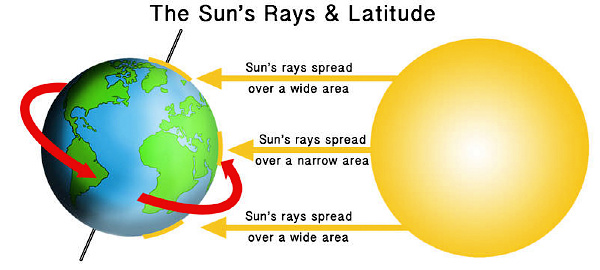
LATITUDE & THE INCIDENCE OF DISEASE: OVERWHELMING PROOF THAT VITAMIN D3 DEFICIENCY CAUSES MOST HUMAN DISEASES (Please note-.…
Before we get started let me just whet your appetite about what is contained in the rest of this article. The results of the most important study on aging EVER, that will be the most important study of aging for all time- have just been released! Steve Horvath’s :
Universal DNA methylation age across mammalian tissues
The study proves conclusively that aging is selected for by evolution and is programmed. A result that contradicts all major mainstream theories of aging that have been proposed since the early 1900’s. It turns out August Weisman got the right answer in 1882 but with the wrong reasoning.
The new study also reveals the true cause of aging at the cellular level- the programmed loss of cellular differentiation.