Key Points
Research suggests Bowles’s APES theory, focusing on Aging, Predation, Extinction, and Sex, may outperform the modern evolutionary synthesis in explaining aging and reproductive strategies.
It seems likely that the APES theory better accounts for programmed aging, lifespan variations based on predation defense, and male sex traits as predator attractants, challenging the modern synthesis’s dominance.
The evidence leans toward the APES theory’s son-king hypothesis for menopause, supported by historical figures like Ramses (93 children) and Genghis Khan (A large percentage of Asian males share his genes), contrasting with the grandmother hypothesis, which Bowles argues is disproven.
An unexpected detail is that the APES theory explains asexual animals in low-predation environments and homosexuality linked to prenatal stress, with studies on rats, mice, and WW2 Germany supporting this.
(Supplements/Hormones Available 90%+ OFF- Vitamin D3, Vitamin K2, DHEA, pregnenolone, melatonin, hyaluronic acid, resveratrol, don’t see it? Just ask). Did…
Abstract
A growing body of evidence challenges the conventional view that aging is merely an accidental byproduct of essential genes and metabolic processes. Instead, this paper revisits a long-overlooked 1998 hypothesis that posited aging is modular—composed of multiple, independently evolved systems that each co-opt the vulnerabilities of the last. Fresh insights are developed concerning short LARP1 (Horvath’s #1 pro-aging gene with an unusual RNA binding site on the protein) a scarcely studied nuclear lncRNA that likely truncates ATM and XP/CS mRNAs and downregulates/prevents the production of WRN by interfering with mRNA spliceosome functions. From these insights, how aging proceeds in at least four evolutionary waves is revealed. System #1 (plant-like vascular/structural decline) appears vestigial in humans, overshadowed/co-opted by Horvath’s universal epigenetic clock. System #2 centers on mitochondrial dysfunction in motile organisms. System #3, tied to advanced DNA repair and immune function, propels progeroid syndromes such as ataxia telangiectasia Cockayne syndrome, and xeroderma pigmentosum. Finally, system #4—emerging alongside sexual reproduction—dominates in Werner’s syndrome, unifying older pathways with newfound genomic instability.
In highlighting short LARP1’s proposed ability to sabotage crucial mRNA splicing leading to defective repair and structural proteins, a surprising synergy is illuminated: these sequentially-evolved senescence pathways act less like random breakdowns and more like a deliberate “orchestra” of aging. Each system is associated with one of the canonical Yamanaka factors (KLF4, Sox2, c-Myc, and Oct4), underscoring the developmental roots of senescence. Far from dismissing aging as a mere trade-off under antagonistic pleiotropy, new evidence is presented consistent with an evolutionarily conserved program—one that likely offers local species-level benefits in predator-rich ecosystems by preserving genetic and phenotypic diversity by preventing excessive, homogenizing contributions to the gene pool by single individuals. The same selection pressure also selects for menopause in humans and declining fertility in animals with aging. Interestingly, the same evolutionary logic that explains aging’s adaptive role applies to the advantage of sexual over asexual reproduction, as sexual reproduction further accelerates genetic (via recombination) and phenotypic diversity and bolsters resilience against evolving predators. For gerontologists, evolutionary theorists, and epigenetic researchers alike, this framework suggests that aging emerges from deeply adaptive, multi-layered processes rather than serendipitous decline, opening avenues for therapeutic disruption and a deeper understanding of life’s final act.
Abstract:
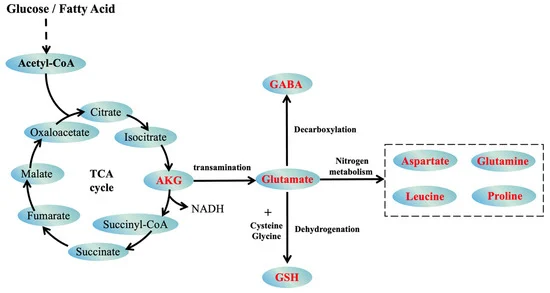
This groundbreaking article presents a unified theory of aging that integrates evolutionary biology, epigenetics, and metabolic regulation, offering a paradigm shift in our understanding of senescence. By synthesizing recent research on Horvath’s epigenetic clock, the GABA-glutamate-αKG axis, and the evolutionary layers of aging systems, the authors propose a compelling model where hormonal changes trigger the expression of short LARP1, a key orchestrator of four distinct aging systems. The theory elucidates how luteinizing hormone (LH), human chorionic gonadotropin (hCG), and follicle-stimulating hormone (FSH) differentially activate these systems, explaining gender-specific aging patterns. Furthermore, it reveals the unexpected role of SP-1 in linking sexual maturation to aging through regulation of MAO-A, MAO-B, and WRN. This comprehensive framework not only explains the acceleration of aging but also identifies novel therapeutic targets, potentially revolutionizing anti-aging interventions. By connecting hormonal changes, metabolic imbalances, and epigenetic dysregulation into a cohesive aging program, this article challenges long-held beliefs about the nature of aging and opens new avenues for extending healthspan and lifespan1.
The remarkable evolutionary studies of Trinidad guppies provide compelling empirical evidence for testing the APES (Aging, Predation, Extinction, and Sex) theory of evolution. This comprehensive analysis examines how David Reznick’s pioneering predator introduction experiments align with the APES framework, offering insights into the dynamic relationship between predation pressure and evolutionary responses in natural populations.
This comprehensive analysis examines how each of Horvath’s 48 epigenetic aging genes may influence magnesium and progesterone metabolism during aging3. The findings reveal multiple direct and indirect pathways through which these genes can disrupt mineral homeostasis and hormone signaling, potentially contributing to age-related deficiencies in both magnesium and progesterone.