Back to the Future:
How Cancer Cells and Stem Cells
Recapture Their Ancestral Past.
Jeff T. Bowles 2/16/2025
Lifespan Bioresearch LLC
Abstract
-
Introduction
Cellular metabolism and structure are critical determinants of cell fate and function. Metazoan cells typically rely on mitochondrial oxidative phosphorylation (OXPHOS) for the majority of their ATP production under aerobic (high oxygen) conditions. However, some highly proliferative cells—such as embryonic stem cells (ESCs) and cancer cells—rely substantially on glycolysis for ATP generation even when oxygen is abundant. This phenomenon, often termed the “Warburg effect,” was first described by Otto Warburg in the 1920s and later formalized in his 1956 paper on respiratory impairment in cancer cells (Warburg, 1956).
Beyond metabolic parallels, ESCs and cancer cells share key structural and genomic features. Notably, lamin A, a key component of the nuclear lamina that regulates chromatin organization and gene silencing, is either absent or expressed at very low levels in both ESCs and many aggressive cancer cells. The lack of lamin A also correlates with a less rigid nucleus, potentially contributing to the plasticity that characterizes stem cells and the invasive capacity of cancer cells.
Moreover, both ESCs and cancer cells exhibit the ability for indefinite self-renewal under the right conditions, reminiscent of simpler ancestral cells that proliferated indefinitely in primordial environments and also similar to modern bacteria and archaea that are immortal. This paper examines the evidence for these parallels, explores the role of apoptosis in protecting multicellular organisms from unregulated proliferation, and discusses the hypothesis that metabolic insufficiencies in mitochondria underlie many aspects of cancer biology.
-
The Warburg Effect in Embryonic Stem Cells and Cancer Cells
- Historical Context
Otto Warburg first observed that cancer cells ferment glucose into lactate even in the presence of oxygen (Warburg, 1956). This “aerobic glycolysis” was paradoxical because oxidative phosphorylation is generally more efficient for ATP production. Subsequent research revealed that highly proliferative cells such as ESCs also rely on glycolysis as their primary energy source (Cho et al., 2006; Zhang et al., 2012). - Mechanistic Underpinnings
- Rapid ATP Turnover and Biosynthesis: Glycolysis can meet the high biosynthetic demands of proliferating cells by providing intermediates for nucleotide, amino acid, and lipid synthesis.
- Hypoxia-Inducible Factors (HIFs): In cancer cells, stabilization of HIF-1α can occur even under normoxia, driving the expression of glycolytic enzymes (Semenza, 2012). In ESCs, a similar mechanism can be triggered, ensuring a primarily glycolytic metabolism (Zhang et al., 2012).
- Evolutionary Perspective
Life forms that predate the oxygenation of Earth’s atmosphere (~2.4 billion years ago) would have relied on anaerobic (oxygen free) metabolism. In that sense, ESCs and cancer cells may recapitulate an ancestral metabolic state, supporting the notion that these cells exhibit “primordial” features.
-
Lamin A, Nuclear Flexibility, and a Primordial Cell State
- Role of Lamin A
- Nuclear Architecture: Lamin A is a key component of the nuclear lamina, providing structural support and silencing genes for proper cell differentiation by interacting with chromatin.
- Gene Regulation: Downregulation or absence of lamin A correlates with changes in chromatin organization and can support a more pluripotent gene expression profile (Constantinescu et al., 2006).
- Lamin A in ESCs
Embryonic stem cells and induced pluripotent stem cells generally have low lamin A expression, allowing for greater nuclear pliability (Eckersley-Maslin et al., 2018). - Lamin A in Cancer
Many aggressive cancer types display reduced lamin A, facilitating the flexibility required for invasion and metastasis (Harouaka et al., 2016). Moreover, the disruption of lamin A function can deregulate normal gene repression, contributing to oncogenic transcriptional profiles.
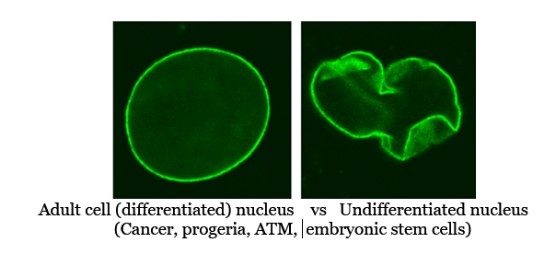
- Ancestral Perspective
Single-celled organisms like bacteria and archaea do not have lamin A, and eukaryotic cells in early evolution likely had simpler nuclear lamina structures. Reduced lamin A in cancer and ESCs might reflect a shift toward a more plastic, developmentally “earlier” cellular state.
-
Immortality and Indefinite Proliferation
- Unlimited Replicative Potential
- ESCs: By definition, ESCs can self-renew indefinitely and give rise to all somatic cell types.
- Cancer Cells: Cancer cells bypass senescence (via telomerase activation or alternative lengthening of telomeres) and can proliferate indefinitely (Shay & Wright, 2011).
- Link to Early Life
Unicellular organisms proliferate as long as nutrients are available. The indefinite proliferation seen in ESCs and cancer cells echoes this property, underscoring the evolutionary argument that they revert to ancestral, single-celled life-like behaviors.
5. Mitosis as a Prerequisite for Apoptosis and Complex DNA Repair
5.1. Evolutionary Perspective: Mitosis Before Apoptosis
Before multicellular life developed sophisticated mechanisms for programmed cell death, single-celled organisms first had to master the process of cell division (mitosis). From an evolutionary standpoint, it is reasonable to propose that mitosis—allowing accurate segregation of genetic material into daughter cells—had to precede the emergence of advanced DNA repair pathways and eventually apoptosis. In other words, cells needed a reliable way to reproduce (mitosis) before developing an equally precise way to eliminate defective cells (apoptosis) (Gottesfeld & Forbes, 1997; Rieder & Khodjakov, 2003).
-
Basic Cellular Propagation
- Early eukaryotic cells required a mechanism to replicate their genetic material and distribute it evenly during cell division.
- Only after this foundational process was in place could more “costly” safeguards such as robust DNA repair mechanisms and programmed cell death evolve to ensure genomic integrity.
-
Foundation for Complex Cell Fate Decisions
- Once mitosis was established, cells that accumulated too much damage—possibly from metabolic byproducts or environmental stress—could develop specialized responses to detect and, if necessary, trigger a self-destruct sequence (apoptosis).
- This evolutionary trajectory aligns with the fact that multicellularity requires strict control over cell populations and that apoptosis emerged as a critical quality-control measure.
5.2. Epigenetic and Transcription Factor Clearance During Mitosis
A key feature of mitosis in modern eukaryotic cells is the widespread reorganization of chromatin and transient removal of transcription factors and many epigenetic marks:
-
Global Transcriptional Shutdown
- During mitosis, the cell halts most gene transcription. This shutdown is partly enforced by the phosphorylation of histone proteins and the eviction of transcription factors from chromatin (Gottesfeld & Forbes, 1997; Kadauke & Blobel, 2013).
- By removing these transcription factors, the cell ensures that no new transcripts interfere with chromosomal segregation.
-
Epigenetic Mark Removal
- Many histone modifications that maintain active or repressed gene states are partially stripped or reconfigured.
- While some “bookmarking” factors persist to help cells “remember” certain gene expression patterns post-division, a significant fraction of epigenetic marks is transiently lost or altered in the process (Kadauke & Blobel, 2013).
-
Vulnerability to DNA Cleavage
- DNA methylation in eukaryotes can help prevent unwarranted nuclease activity (though the exact parallels to prokaryotic restriction-modification systems are more nuanced). If a hypothetical or aberrant endonuclease were present at high levels while epigenetic protections were relaxed, the DNA could become more susceptible to cleavage (Negishi et al., 2014).
- This momentary exposure reflects a primordial risk, which in early evolutionary contexts might have driven the development of more robust DNA repair or protective mechanisms to guard the genome during cell division.
5.3. Linking Mitosis to Apoptosis
During later evolutionary stages, cells combined these mitotic clearance processes with emerging DNA repair pathways. When DNA damage or metabolic insufficiency surpassed a critical threshold, the cell could pivot from attempting mitosis or repair to initiating apoptosis. Hence, in modern cells:
- Mitotic Checkpoints constantly assess DNA integrity before proceeding with division (Rieder & Khodjakov, 2003).
- If severe damage is detected—and energy resources permit—the cell may activate apoptotic pathways instead of completing mitosis (Green & Kroemer, 2004).
- This evolutionary layering (mitosis → DNA repair → apoptosis) highlights how each system builds upon the last, reflecting the cell’s dual goals of reproduction and survival.
6. Apoptosis: Returning to a Primordial State Before Death?
- Mechanism of Apoptosis
Apoptosis is a programmed cell death pathway crucial for eliminating damaged, infected, or unneeded cells. During apoptosis:- The cell undergoes nuclear condensation (pyknosis).
- Caspases orchestrate the cleavage of key cellular proteins.
- DNA is fragmented into nucleosomal units.
- Demethylation and Loss of Transcription Factors
- Demethylation: Evidence suggests that in the final stages of apoptosis, DNA can undergo widespread demethylation (Makarovsky et al., 2020). If true across many cell types, this would briefly revert chromatin to a more “open” or permissive state.
- Transcription Factor Loss: The proteolytic activity of caspases can degrade multiple proteins, including transcription factors, although the complete clearance of all transcription factors is still under investigation (Luthi & Martin, 2007). This leads to a shutdown of gene Makarov sky suppression/expression programs returning the cell to an undifferentiated pluripotent state.
- Convergence on a “Primordial” State
The hypothesis here is that by removing epigenetic marks and transcription factors prior to fragmentation, the cell transiently returns to a more primitive epigenetic landscape. This stands in stark contrast to the typical role of apoptosis in multicellular organisms: to ensure that such a cell is destroyed rather than allowed to remain immortal
7. Cancer as a Failure of Apoptosis
- Stalled Program
The theory suggests that cancer results when the apoptosis program fails midway, leaving the cell in a proliferative state that still retains features of a more “stem-like” or “primordial” metabolism.- Insufficient Mitochondrial Energy: Mitochondria are vital not only for ATP production but also for apoptotic signaling through the release of cytochrome c. If mitochondrial dysfunction compromises ATP levels, the final cleavage of DNA and irreversible steps of apoptosis may not complete, leaving the cell alive but dysregulated (Green & Kroemer, 2004).
- Metabolic Basis of Failure
Thomas Seyfried’s metabolic theory of cancer posits that impaired mitochondrial function is a key driver of oncogenic transformation (Seyfried et al., 2014). Because energy-dependent steps in apoptosis and DNA repair are compromised, cells default to a glycolytic, rapidly proliferating phenotype. - Evidence from Mitochondrial Restoration
- Cancer Cell Reversion: Experiments in which functional mitochondria are introduced into cancer cell lines have shown that normal respiratory function can sometimes be restored, along with growth suppression or restored susceptibility to apoptosis (Kaipparettu et al., 2013).
- Embryo Formation: There are reports (though still controversial) suggesting that cancer cell nuclei can support normal embryogenesis when fused with oocytes that contain healthy mitochondria, indicating that defective mitochondria—and not the nucleus alone—play a critical role in tumorigenicity (Cervera et al., 1974; Illmensee & Mintz, 1976).
8. Common Oncogenic Mutations That Impair Apoptosis
An array of well-characterized oncogenic mutations can promote cancer cell survival by disrupting the tightly regulated apoptotic machinery. In many cases, these mutations converge on pathways that become especially critical when ATP levels are low, thwarting the cell’s normal ability to undergo programmed cell death and instead favoring proliferation. Below is a more detailed analysis of how mutations in several key genes—TP53, RB1, PTEN, MYC, the BCL-2 family, and BRCA2—compromise apoptosis and drive tumorigenesis.
- TP53
TP53 is often called the “guardian of the genome” due to its central role in detecting cellular stress (e.g., DNA damage, metabolic insufficiency) and orchestrating either cell-cycle arrest or apoptosis (Kastan et al., 1991; Freed-Pastor & Prives, 2012). When TP53 is mutated, cancer cells lose a critical checkpoint that normally halts cell division in the face of DNA lesions or low ATP availability. Functionally, p53 initiates apoptosis by upregulating pro-apoptotic genes such as BAX, PUMA, and NOXA while simultaneously repressing pro-survival signals. In conditions of insufficient energy, cells with functional p53 typically undergo apoptosis rather than continuing to proliferate with compromised ATP levels. However, p53-deficient cells evade this fate by failing to activate the downstream caspase cascade, allowing them to survive despite metabolic stress. - RB1 (Retinoblastoma 1)
RB1 encodes the retinoblastoma protein (pRB), which classically regulates the G1/S transition in the cell cycle by controlling the activity of E2F transcription factors (Sherr, 2004). In addition to cell-cycle control, intact RB1 can influence apoptosis through several mechanisms—for example, by repressing E2F-mediated expression of genes that drive proliferation in metabolically stressed cells. Loss-of-function mutations in RB1 result in unchecked E2F activity, enhancing cell division even under conditions where ATP levels and other resources would normally limit proliferation. Moreover, free E2F can induce the expression of anti-apoptotic factors or interfere with pro-apoptotic signaling, thereby blunting the cell’s ability to initiate programmed cell death when energy supplies wane. - PTEN (Phosphatase and Tensin Homolog)
PTEN negatively regulates the PI3K/AKT pathway, which is a major pro-survival and growth signaling cascade in cells (Maehama & Dixon, 1998). In healthy cells, PTEN dephosphorylates phosphatidylinositol (3,4,5)-trisphosphate, thereby limiting AKT activation. When PTEN is lost or mutated, AKT remains chronically active, promoting increased glycolysis (and, indirectly, the Warburg effect), cell survival, and inhibition of apoptosis (Eng, 2003). Elevated AKT can directly phosphorylate and inactivate multiple pro-apoptotic proteins (including BAD), thereby buffering cells from death signals even under low ATP conditions. This aberrant survival advantage is especially problematic when mitochondrial energetics are compromised, as the cell may bypass the normal metabolic checkpoints that would trigger apoptosis. - MYC
MYC is a transcription factor that drives cell proliferation, growth, and metabolism (Dang, 2012). While overexpression of MYC can paradoxically sensitize cells to apoptosis (when pro-apoptotic signals remain intact), MYC-driven tumors often acquire additional mutations (e.g., in TP53 or BCL-2 family members) that thwart apoptotic pathways (Adhikary & Eilers, 2005). In metabolic terms, MYC upregulates glycolytic enzymes and glutamine metabolism, fostering an environment where cancer cells can adapt to fluctuating ATP availability. If critical pro-apoptotic checkpoints (e.g., p53-mediated) are simultaneously disabled, MYC-overexpressing cells can continue to divide despite inadequate oxidative phosphorylation, further compromising the cell’s intrinsic ability to undergo energy-dependent apoptosis. - BCL-2 Family (BCL-2, BCL-XL, MCL-1, etc.)
Members of the BCL-2 family tightly govern the mitochondrial (intrinsic) pathway of apoptosis. Anti-apoptotic proteins such as BCL-2, BCL-XL, and MCL-1 inhibit the release of cytochrome c and other pro-apoptotic factors from mitochondria (Cory et al., 2003). By contrast, their pro-apoptotic counterparts (BAX, BAK, BID) promote mitochondrial outer membrane permeabilization (MOMP), enabling caspase activation. Mutations or overexpression of BCL-2 and related genes tip the balance toward survival, making cells far more resistant to energy stress-induced apoptosis. In such a state, even if ATP levels drop, mitochondrial outer membrane permeabilization is prevented or delayed, impeding the final steps of apoptosis and potentially leading to the “stalled” apoptotic phenotype described in metabolic theories of cancer. - BRCA2
BRCA2 primarily functions in homologous recombination repair of DNA double-strand breaks (Venkitaraman, 2002). While BRCA2 mutations are best known for driving genomic instability, these defects also intersect with apoptotic pathways. Persistent DNA damage normally triggers p53-dependent or p53-independent apoptosis; however, in a BRCA2-mutant background, cells often accumulate additional mutations in TP53 or other checkpoint regulators (Roy et al., 2011). This cumulative damage further compromises the apoptotic response. When insufficient ATP availability compounds the problem—because high-fidelity DNA repair and the final stages of apoptosis both require energy—BRCA2-deficient cells can escape apoptosis, continuing to propagate mutations that fuel tumor progression.
By altering critical checkpoints and signaling cascades, these common genetic mutations effectively short-circuit apoptosis, particularly under metabolic stress. The result is a cell population capable of continued proliferation even when ATP supplies wane, thus setting the stage for unchecked growth and tumor evolution.
Future Perspectives and Research Directions
- Refining the Epigenetic Model of Apoptosis
- Extent of Genome-Wide Demethylation: More quantitative studies are needed to map the extent of DNA demethylation in apoptotic cells and to determine whether transcription factors are consistently depleted.
- Single-Cell Approaches: Single-cell multi-omics could provide a powerful way to track epigenetic and transcriptional changes from the earliest stages of apoptosis to the final DNA fragmentation.
- Mechanisms Underlying Lamin A Loss
- Regulation in Stem vs. Cancer Cells: Determining whether similar or divergent pathways downregulate lamin A in ESCs and cancer cells could illuminate fundamental nuclear structure-function relationships.
- Therapeutic Implications
- Metabolic Therapies: Interventions to boost mitochondrial function (ketogenic diets, oxidative phosphorylation enhancers, or direct mitochondrial transfer) might push cancer cells toward completing apoptosis or reverting to non-tumorigenic phenotypes.
- Targeting Mitochondrial Biogenesis: Drugs that promote healthy mitochondrial biogenesis may correct metabolic defects in cancer cells and restore their susceptibility to cell death cues.
- Evolutionary Medicine
- Cancer as Atavism: The concept of cancer cells reverting to a unicellular, ancestral metabolic state invites further research in evolutionary biology. Investigating how and why these pathways might re-emerge offers a unique lens on cancer prevention and treatment.
-
Conclusion
The parallels between embryonic stem cells and cancer cells—including their reliance on fermentation-based metabolism (Warburg effect), downregulation of lamin A, and capacity for indefinite proliferation—highlight a compelling view of cancer as a reversion to a more primitive, single-celled life form. Coupled with the observation that apoptosis involves genome-wide epigenetic changes that temporarily restore a stem-like state prior to cell death, it becomes evident how metabolic dysfunctions in mitochondria could stall cell death and promote unregulated proliferation.
Thomas Seyfried’s hypothesis that cancer is primarily a metabolic disease finds resonance with these observations. Restoring robust mitochondrial function—through metabolic or genetic interventions—may tip the balance either toward normal cellular homeostasis or toward effective apoptosis. While many questions remain, these lines of inquiry open promising avenues for new therapeutic approaches and offer a deeper evolutionary understanding of cancer biology.
References
- Cervera, M., et al. (1974). Demonstration of the oncogenic potential of cells derived from a mouse teratocarcinoma in normal blastocysts. Nature, 248, 678–681.
- Cho, Y. H., et al. (2006). Functional bioenergetic analysis of human embryonic stem cells and embryoid bodies reveals the importance of oxidative phosphorylation for cardiomyocyte differentiation. J Biol Chem, 281, 37307–37319.
- Constantinescu, D., et al. (2006). Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells, 24, 177–185.
- Eckersley-Maslin, M. A., et al. (2018). Epigenetic priming by Dppa2 and Dppa4 in pluripotency facilitates multi-lineage commitment. Nat Struct Mol Biol, 25, 110–122.
- Green, D. R. & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305, 626–629.
- Harouaka, R., et al. (2016). Low lamin A levels enhance tumor cell dissemination and metastasis by releasing the nuclear envelope pressure on invasive genes. Cancer Res, 76, 4906–4917.
- Illmensee, K., & Mintz, B. (1976). Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A, 73, 549–553.
- Kaipparettu, B. A., et al. (2013). Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS One, 8, e61753.
- Luthi, A. U., & Martin, S. J. (2007). The CASBAH: a searchable database of caspase substrates. Cell Death Differ, 14, 641–650.
- Makarovsky, D., et al. (2020). Global DNA demethylation triggers ESC-like gene expression in apoptotic mouse embryonic fibroblasts. Sci Rep, 10, 13422.
- Semenza, G. L. (2012). Hypoxia-inducible factors in physiology and medicine. Cell, 148, 399–408.
- Seyfried, T. N., et al. (2014). Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis, 35, 515–527.
- Shay, J. W., & Wright, W. E. (2011). Role of telomeres and telomerase in cancer. Semin Cancer Biol, 21, 349–353.
- Warburg, O. (1956). On respiratory impairment in cancer cells. Science, 124, 269–270.
- Zhang, J., et al. (2012). Metabolism in pluripotent stem cells and early mammalian development. Cell Metab, 15, 324–331.Adhikary, S., & Eilers, M. (2005). Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol, 6, 635–645.
- Gottesfeld, J. M., & Forbes, D. J. (1997). Mitotic repression of the transcriptional machinery. Trends Biochem Sci, 22(1), 13–18.
- Green, D. R., & Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science, 305(5684), 626–629.
- Kadauke, S., & Blobel, G. A. (2013). Mitotic bookmarking by transcription factors. Epigenetics Chromatin, 6(1), 6.
- Negishi, M., et al. (2014). eEF1BδL is involved in the post-transcriptional regulation of embryonic stem cell self-renewal and differentiation. Sci Rep, 4, 4260. (Cited for illustrative mention of epigenetic vulnerabilities; actual data pertains to ESC regulatory factors.)
- Rieder, C. L., & Khodjakov, A. (2003). Mitosis through the microscope: advances in seeing inside live dividing cells. Science, 300(5616), 91–96.
- Cory, S., et al. (2003). Bcl-2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer, 3, 371–382.
- Dang, C. V. (2012). MYC on the path to cancer. Cell, 149, 22–35.
- Eng, C. (2003). PTEN: one gene, many syndromes. Hum Mutat, 22, 183–198.
- Freed-Pastor, W. A., & Prives, C. (2012). Mutant p53: one name, many proteins. Genes Dev, 26, 1268–1286.
- Kastan, M. B., et al. (1991). Participation of p53 protein in the cellular response to DNA damage. Cancer Res, 51, 6304–6311.
- Maehama, T., & Dixon, J. E. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem, 273, 13375–13378.
- Roy, R., et al. (2011). BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer, 12, 68–78.
- Sherr, C. J. (2004). Principles of tumor suppression. Cell, 116, 235–246.
- Venkitaraman, A. R. (2002). Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell, 108, 171–182.