Evolution’s Suicide Switch:
MAO-B Forces a Rethink of Darwin’s Legacy
Dr. Eric Berg, Tim Ioannides MD, Stylianos Jordanis Phd, Albert Wahlström, Jeff T. Bowles
Lifespan BioResearch LLC
Contact information: jbowles1984@kellogg.northwestern.edu
1/06/2025
Abstract
Monoamine Oxidase A (MAO-A) and Monoamine Oxidase B (MAO-B) are flavin-dependent enzymes that progressively increase with age in many tissues. It is proposed that both serve as “death genes,” depleting Flavin Adenine Dinucleotide (FAD) and thereby reducing mitochondrial energy production—mirroring the known action of CD38, which depletes Nicotinamide Adenine Dinucleotide (NAD+). Although MAO-A retains certain developmental and sex-related roles, MAO-B appears to confer no clear early-life benefit and emerges as the first true, fully dedicated death gene documented. This discovery challenges classical evolutionary theories and suggests an unexpected “programming” of aging. The contrasting knockout phenotypes are detailed—dramatic aggression and neurotransmitter imbalance for MAO-A vs. subtle or minimal deficits for MAO-B. How the parallel depletion of NAD+ (by CD38) and FAD (by MAOs) undermines electron transport chain function in a near-symmetric manner is also examined. These findings open new therapeutic possibilities, including targeted inhibition of MAO-B (and MAO-A) and combined strategies preserving both NAD+ and FAD to mitigate age-related decline. This finding also calls into question a core principle of the selfish gene theory of evolution and suggests a need for a reevaluation of mainstream theory.
Introduction
The possibility that certain genes exist solely to promote aging and death has long been considered anathema to classical evolutionary theory. Typically, “antagonistic pleiotropy” or “disposable soma” models hold that late-life deleterious genes persist only because they confer early-life advantages (1) (2). Recently cracks have started to appear in the veneer of this model of evolution where “nothing deleterious to individual fitness such as aging could evolve for the benefit of the group”. The biggest challenge to the impossibility of the selection for aging recently emerged with Steve Horvath’s seminal study showing that mammals share a universal aging system that is highly conserved and thus by inference was selected for by evolution (3). Interestingly one of the genes found to be highly associated with aging in Horvath’s paper is SP-1. SP-1, a transcription factor, regulates the expression of both MAO-A and MAO-B genes by binding to their promoter regions, enhancing MAO-A transcription and either activating or repressing MAO-B depending on other factors.
What follows might be the final nail in the coffin of the idea that it is impossible for evolution to have selected for aging and thus for death genes, whose sole purpose is to kill at an older age.
A novel observation has been made that dramatic increases in the FAD-dependent enzymes Monoamine Oxidase A & B (MAO-A, MAO-B) in many tissues with aging might deplete FAD from the electron transport chain (ETC) in a manner similar or parallel to how CD38 increases deplete NAD+. While this mechanism of FAD depletion if it exists, would contradict or require reexamination of mainstream thinking about FAD metabolism during aging, it would provide an eloquent and simple mirror image of the aging dynamics seen in the NAD+/CD38 aging system.
The aging dynamics seen in the interaction between CD38 and NAD+ during aging include large increases in various tissues of CD38 levels caused by increased production by M1 (the inflammatory) macrophages. Increased CD38 destroys NAD+ when it uses it as a cofactor. With aging CD38 increases by as much as 50% in the in the ilium, jejunum, and kidney, CD38 NADase activity increased by at least 50% during the aging process in in the ilium, jejunum, and kidney (4).Larger increases of 2.0 to 2.5x are seen in many other tissues.
The depletion of FAD by enzymes dependent on it have not been considered a s a cause of aging by mainstream scientists by MAO due to the nature of how FAD and MAO’s interact. Rather than being destroyed by MAO enzymes, FAD is thought to be recycled, and thus it is assumed the pool of FAD in the mitochondria does not decline due to increases in MAOs in a manner similar to the pool of NAD+ caused to decrease by CD38 increases. This this idea is an unstudied area of aging research.
Now what if there is something wrong with the current understanding of how MAO s and FAD interact? What if somehow Mao increases were also decreasing FAD pools similar to the manner in which CD38 decreases NAD+ pools, well let’s then assume this is the case- we will examine the possibilities later. And now let’s take a look at what we would have. It sure looks very elegant symmetric and almost a perfect mirror image of the CD38/NAD+ aging system. Consider-
NAD+ and FAD are each responsible for 40% of the protons produced by the electron transport chain (ETC) .
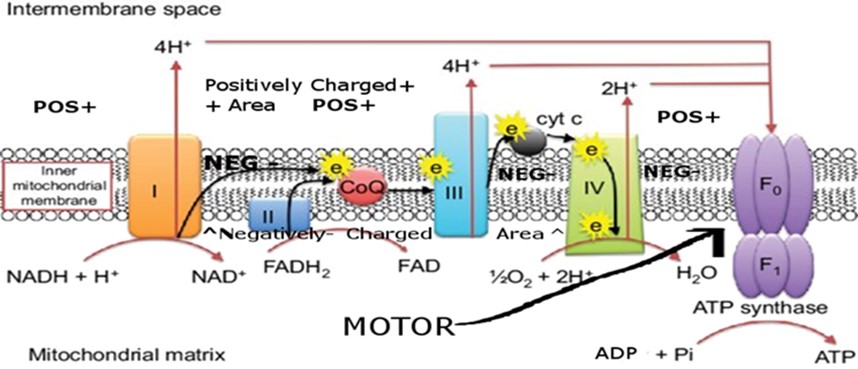
Interestingly, Monoamine Oxidase A (MAO-A), which evolutionarily diverged from the more ancient MAO-B, retains partial utility in controlling aggression, mood, and sexual/reproductive behavior, and also has roles in development during early life. However, both MAO-A and MAO-B are FAD-dependent and are proposed herein to drive aging by consuming FAD as they escalate with age in tissues such as the heart and brain and many others (4) (5).
Further heightening the significance of this phenomenon is both MAOs mirror-like parallel to CD38, an enzyme that increases with age to deplete NAD+—another essential electron carrier. NAD+ and FAD are almost structurally identical except for their distinct vitamin moieties (B3 vs. B2); each contributes roughly 40% of the electron transport chain’s proton pumping capacity. As such, combined declines in NAD+ and FAD can undermine up to 80% of mitochondrial energy output in older organisms, fueling oxidative stress, organ dysfunction, and the classic signs of aging.
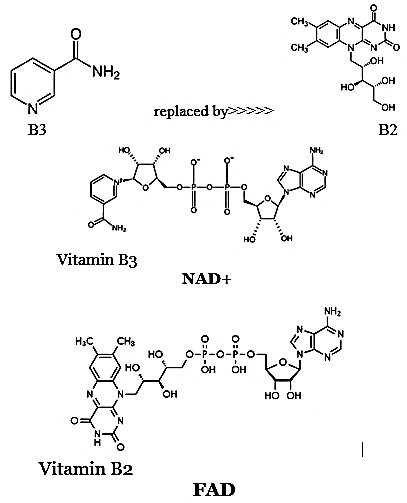
Structural Similarity Between NAD+ and FAD
Common Features
Both NAD+ (Nicotinamide Adenine Dinucleotide) and FAD (Flavin Adenine Dinucleotide) share several core structural components:
- Adenine base: Each molecule contains an adenine nucleotide.
- Ribose sugar: The adenine attaches to a ribose sugar, forming a nucleoside.
- Phosphate groups: Both coenzymes incorporate phosphate groups linking subunits.
- Redox capabilities: Each coenzyme can accept and donate electrons, enabling redox reactions crucial for cellular respiration.
The Vitamin Moieties
The key distinction lies in the vitamin-derived section:
- NAD+: Contains niacin (vitamin B3) as its reactive nicotinamide group.
- FAD: Contains riboflavin (vitamin B2) in place of nicotinamide.
Because these molecules are otherwise so structurally and functionally similar, each supports roughly 40% of the electron transport chain’s proton pumping capacity. Thus, a deficit in either NAD+ or FAD severely compromises mitochondrial ATP generation.
MAO-A and MAO-B as Flavin-Dependent (FAD) Death Genes
Because MAO-B appears to serve no beneficial function in early life but rather only profound detrimental effects in older age, it raises the provocative hypothesis that it is a true death gene—one whose age-related surge serves no beneficial function while promoting senescence by depleting Flavin Adenine Dinucleotide (FAD) in mitochondria.
Evolutionary Background
MAO-B is believed to have evolved first, later undergoing duplication to yield MAO-A. Over millions of years, MAO-A acquired beneficial or at least pleiotropic tasks (e.g., aggression control, sexual behavior, some developmental roles), while MAO-B appears increasingly specialized as an enzyme that surges in older age, with no clear benefit but potent and numerous detrimental effects.
Shared Mechanism: FAD Consumption
Both MAO-A and MAO-B:
- Require FAD as a cofactor to deaminate monoamines (dopamine, serotonin, etc.)
- Rise significantly with aging in many tissues, notably heart and brain.
- Potentially deplete or sequester FAD, limiting its availability for Complex II in the electron transport chain.
The net result is a progressive decline in mitochondrial efficiency, analogous to CD38’s depletion of NAD+ in older organisms.
Tissue-Specific Rises
- Brain: MAO-B may increase by several hundred percent (~400%) with age, while MAO-A often increases more modestly (e.g., ~34% / 16% in perimenopausal / postmenopausal women’s brains, but can be higher in certain pathologies). MAO-A also fluctuates on a monthly cycle in females, notably, being inhibited by estrogen and promoted by progesterone (6). MAO-A inhibitors were the first anti-depressants used in medicine beginning in the 1950’s (7) and are still in limited use today.
- Heart: Both MAO-A and MAO-B can climb by 400–600% in the aging heart, aggravating oxidative stress, myocardial dysfunction, and heart failure.
- Other Tissues: Pineal gland, kidney, liver and many others also exhibit elevated MAO isoforms in older age, correlating with age-related pathologies. A quick search of the online Human Protein Atlas https://www.proteinatlas.org/search/MAO will show you just how pervasive MAO is expressed in all the various tissues.
MAO-A Knockout vs. MAO-B Knockout
MAO-A Knockout: Dramatic Behavioral Effects
- Elevated Aggression: MAO-A−/− male mice show extreme aggression, impulsivity, as well as “autistic-like” traits (8). A Dutch family that suffered form a nonfunctional MAO-A gene produced male offspring with exceptional criminal tendencies. One son tried to run his boss over with a car, another tried to kill the prison warden with a pitchfork and a third raped his sister and was an arsonist. The nonfunctional MAO-A gene did not affect a female family member as much possibly because it is an X linked gene meaning she may have benefited from a functional copy (9).
- Neurotransmitter Overflow: Knockout leads to high levels of serotonin, norepinephrine, and dopamine, revealing MAO-A’s pivotal role in life behavior modulation (10).
- Developmental Impact: Absence of MAO-A during critical windows can cause severe, lasting neurobehavioral abnormalities (11).
MAO-B Knockout: Minimal or Subtle Changes
- Mild Anxiety/Behavioral Shifts: Some studies note slight decreases in anxiety or mild risk-taking increases (12).
- Elevated Phenylethylamine (PEA): Without MAO-B, PEA accumulates, imparting a mild stimulant effect—yet with limited overt pathology (13).
- Protective Effects:
- Reduced oxidative stress in models of heart failure (14).
- Possible prostate cancer amelioration in certain models (15).
- Some neuroprotective actions (e.g., resistance to neurotoxins in Parkinson’s models, complete protection from Parkinson’s disease in mouse models) (16).
Such minimal deficits in MAO-B−/− animals underscore the notion that MAO-B does not serve critical early-life functions—unlike MAO-A, whose knockout phenotype is significant. This aligns with MAO-B being more purely dedicated to harm in late life, strongly supporting its status as a true death gene.
MAO-A vs. MAO-B: Degrees of ‘Death Gene’ Status
- MAO-A:
- Increases significantly with aging in many issues (especially the heart, some brain regions)
- Still retains beneficial or at least indispensable developmental/behavioral roles (sex differences, mood, aggression control).
- Hence, it is only partly a “death gene” by virtue of its late-life surge, but does not fully fit the “only harmful” criterion.
- MAO-B:
- Surges dramatically (100s of percent) in multiple organs with minimal baseline benefit.
- Knockout is nearly inconsequential until old age, implying no trade-off advantage.
- Hence, stands out as the first known example of a 100% dedicated death gene—no recognized early utility, purely detrimental in old age.
Mirror Mechanisms:
CD38 / NAD+
vs.
MAO-A & MAO-B / FAD
Parallel Increases with Age
- CD38: Expression/activity escalates with aging, leading to NAD+ depletion, reduced sirtuin activity, and metabolic collapse (17).
- MAO-A and MAO-B: Increase in tandem, consuming FAD in the same mitochondrial environment (18).
Shared Consequences
- Mitochondrial Energy Depletion:
- NAD+ and FAD each power ~40% of the ETC’s proton pumping.
- Combined shortfalls can cause up to an ~80% reduction in ATP generation.
- Oxidative Stress:
- CD38-mediated NAD+ depletion impairs sirtuins, raising ROS (19).
- MAO-mediated FAD depletion triggers electron leakage, plus direct hydrogen peroxide production from monoamine deamination (20) (21).
- Age-Related Diseases:
Below is a concise, data-driven comparison of the diseases linked to elevated CD38, MAO-A, and MAO-B, drawing on available research in the scientific literature (journal articles, reviews, and reputable online databases). All three enzymes, when overexpressed with aging, can drive mitochondrial dysfunction and oxidative stress—yet each has particular disease emphases depending on tissue distribution and substrate specificity.
Overview of CD38, MAO-A, and MAO-B
CD38
- Primary Function: NAD+ glycohydrolase, depleting NAD+ and impairing sirtuins and mitochondrial function.
- Tissue Expression: Prominently expressed in immune cells (B and T lymphocytes), also found in cardiac tissue, adipose tissue, and the nervous system.
- Age-Related Elevation: Increases with age across various tissues, correlating with systemic NAD+ decline.
- CD38 elevation may be caused in part by increasing M2 to M1 macrophage conversion triggered by declining serum GABA and AKG levels with aging (author’s hypothesis).
MAO-A & MAO-B
- Primary Function: FAD-dependent monoamine oxidases that deaminate neurotransmitters and amines, producing H2O2 (and thus oxidative stress).
- Tissue Expression: Both are found on the outer mitochondrial membrane but exhibit different patterns:
- MAO-A is abundant in brain regions regulating mood, aggression; also, in cardiac, intestinal, and other tissues.
- MAO-B is highly expressed in astrocytes and many peripheral tissues (platelets, heart, etc.).
- Age-Related Elevation: Both rise in many tissues with aging—MAO-B sometimes by several hundred percent (e.g., in the heart and brain), and MAO-A significantly in the heart (several hundred percent) and to a lesser extent (34%) in perimenopausal women’s brains and (16%) in postmenopausal brains (22).
Key Differences
- Localization:
- CD38 is found on cell surfaces and within organelles (nucleus, etc.).
- MAOs localize to the outer mitochondrial membrane.
- Substrate Specificity:
- CD38 targets NAD+, NMN, other NAD+ precursors.
- MAOs catabolize monoamines, using (and thus depleting) FAD.
- Evolutionary History:
- CD38 arises in immune regulation contexts.
- MAO-B predates MAO-A, which later gained some beneficial roles.
Despite these differences, both systems converge on the core phenomenon of age-linked cofactor depletion, crippling mitochondrial respiration and accelerating senescence.
Similarities in Disease Associations
Neurodegenerative Diseases
- CD38:
- Implicated in Alzheimer’s disease, as NAD+ depletion and defective sirtuin activity can promote amyloid pathology and neuronal metabolic decline (23).
- Some studies link higher CD38 expression on microglia/immune cells in the CNS to neuroinflammation (24).
- MAO-A:
- Elevated in some Alzheimer’s and Huntington’s contexts (25).
- Can contribute to neuronal damage through hydrogen peroxide production and pro-inflammatory signaling (26).
- MAO-B:
- Strongly linked to Parkinson’s (excess astrocytic MAO-B fosters dopaminergic neuron stress) (27).
- Also implicated in Alzheimer’s disease pathogenesis, particularly in astrocyte-mediated oxidative injury (28).
- Also linked to periodontal disease suggesting the connection between tooth decay and Alzheimer’s as described in the literature (29).
Cardiovascular & Heart Disease
- CD38:
- Overexpression can exacerbate cardiac dysfunction, as NAD+ depletion impairs mitochondrial ATP production in cardiomyocytes (30).
- Associated with heart failure, arrhythmias, and hypertension in aging models (31).
- MAO-A:
- Known to rise significantly in aged hearts (often +400–600%) (32).
- Produces local ROS and aldehydes that damage cardiomyocytes, contributing to ventricular dysfunction and heart failure (33).
- MAO-B:
- Also implicated in cardiac aging; increased MAO-B activity leads to oxidative stress, mitochondrial depolarization, and tissue remodeling (34).
Cancer
- CD38:
- Highly expressed in some hematological malignancies (e.g., multiple myeloma) and can shape tumor microenvironment by modulating NAD+ and inflammatory signals (35).
- Elevated in certain solid tumors, but best recognized in immune-related malignancies (36).
- MAO-A:
- Overexpression noted in lung, breast, prostate, and gastric cancers (37).
- Enhances tumor aggressiveness by inducing oxidative stress, promoting epithelial-mesenchymal transition, and altering local microenvironment (38).
- MAO-B:
- Elevated in prostate cancer stroma and other tumors (39).
- Contributes to tumor progression (e.g., influencing fibroblast or astrocyte cross-talk, generating hydrogen peroxide) (40).
(The common denominator of impaired mitochondrial function and energy output in cancer in all three of the above cases adds weight to the validity of Thomas Seyfried’s idea of cancer as a metabolic / mitochondrial disease as opposed to a being a genetic / mutation disease. (41))
Metabolic Syndrome & Obesity
- CD38:
- Well-documented role in metabolic disorders; increased CD38 activity in adipose tissue correlates with NAD+ decline and insulin resistance (42).
- Linked to obesity, type 2 diabetes, and chronic inflammatory states (43).
- MAO-A:
- Emerging evidence ties it to obesity and insulin resistance; less extensively characterized than CD38 in metabolic disease but still implicated in systemic inflammation and lipid metabolism (44).
- MAO-B:
- Growing research suggests involvement in metabolic disturbances, especially where chronic inflammation and mitochondrial dysfunction intersect. Also, studies are emerging that show that altering MAO-A & B levels can lead to weight loss without a reduction in food intake by resetting the weight set point (45) (46).
Chronic Inflammation & Immune Dysregulation
- CD38:
- Central to “inflammaging.” Overexpressed on immune cells, fueling NAD+ loss and impeding beneficial NAD+-dependent processes (47).
- Contributes to pathologies of autoimmunity and low-grade chronic inflammation in older adults (48).
- MAO-A & MAO-B:
- Less direct roles in immune cell function; however, they do produce reactive byproducts (H2O2, aldehydes) that drive local or systemic inflammation (49).
- Elevated in various tissues under inflammatory stress, e.g., astrocytes (MAO-B), microglia (MAO-A/B) (50).
Comparative Table
| Disease/Condition | Elevated CD38 | Elevated MAO-A | Elevated MAO-B |
| Neurodegeneration | Alzheimer’s, NAD+ depletion → neuronal stress | Alzheimer’s, Huntington’s (moderate evidence); ROS damage | Parkinson’s (strong link), Alzheimer’s (astrocyte involvement) |
| Heart Disease | Heart failure, arrhythmias (NAD+ deficiency) | Ventricular dysfunction, remodeling (FAD depletion, ROS) | Similar to MAO-A (cardiac aging, oxidative stress) |
| Cancer | Multiple myeloma, some solid tumors (immune angle) | Breast, lung, prostate, gastric (tumor aggression, EMT), | Prostate stroma, glioma (tumor microenvironment, fibroblast cross-talk) |
| Metabolic Syndrome | Obesity, insulin resistance, type 2 diabetes | Possibly obesity, insulin resistance (fewer direct studies) | Growing association with obesity, synergy with A or alone |
| Chronic Inflammation | Inflammaging; immune cell overactivation | Produces local ROS, synergy with tissue inflammation | Produces local ROS (esp. astrocytes), fosters proinflammatory damage |
| Immune Dysregulation | Strong (B/T lymphocytes, plasma cells) | Less direct role in immune cells | Less direct role (platelets, astrocytes, not primarily immune) |
| Behavioral/Psych. | Minimal direct | Major (aggression, mood changes, postpartum depression) | Mild (PEA accumulation in knockout, some anxiety changes) |
Important Points-
Overlap in Disease Profiles
- Neurodegeneration: Alzheimer’s for CD38 and MAO-A/B; Parkinson’s strongly tied to MAO-B.
- Heart Disease: All three heavily implicated via mitochondrial energy depletion (NAD+ or FAD).
- Cancer: Each can drive tumor progression, albeit CD38 is often pivotal in hematological malignancies, while MAO isoforms feature in solid tumors (notably MAO-B in prostate cancer).
Unique Nuances
- CD38 → robust immune cell involvement, including multiple myeloma and autoimmunity.
- MAO-A → critical for mood, aggression, postpartum/menopausal depression; upregulated in heart disease and certain cancers.
- MAO-B → minimal early-life function (knockout is well-tolerated), major role in astrocytic and neuronal damage, strongly implicated in Parkinson’s.
Common Mechanistic Thread
- Elevated Enzyme Activity + Cofactor Depletion = amplified oxidative stress, mitochondrial dysfunction, and fueling of chronic inflammatory or degenerative conditions.
By recognizing these shared and distinct disease patterns, we see that CD38, MAO-A, and MAO-B are all crucial age-related enzymes whose overactivity leads to significant metabolic and cellular stress. Each enzyme’s emphasis in different tissues (immune vs. neurological vs. cardiac) shapes how these pathologies manifest, yet the net effect—energy crisis, ROS generation, and organ decline—illustrates their mirror-like roles in aging biology.
Implications for Evolutionary Theory
- Contradiction of Traditional Models:
Genes that reduce fitness in old age typically require an early-life benefit to persist. MAO-B, however, shows no known early advantage. - Possible Explanations:
- Higher Level Selection: Older individuals are removed for population benefit.
(This is most likely in the author’s opinion—species (not group) selection: aging/sexual species (diverse) survive while non-aging/asexual species (identical) go extinct in local environments subject to evolving predation. Aging/sexual species then migrate elsewhere and take over niches left empty by other extinct nonaging/asexual species. (i.e., the aging/sexual rabbit of one ecosystem migrates to replace the extinct non-aging/ asexual ground squirrel of another ecosystem). Result—Selection for aging and sex is occurring almost everywhere all the time. Benefit of aging and sex? The increase and preservation of genetic/phenotypic diversity which allow a faster evolution of defense to evolving predation. This model also allows the existence of non-aging and asexual organisms and also explains why they are rare while selfish gene theory would predict they should be ubiquitous. Some readers may be surprised at some of the animals that can reproduce asexually such as some fish and sharks, California condors, Komodo dragons, boa constrictors, rattlesnakes and even turkeys and chickens. And completely asexual reproducers include whiptail lizards, cave crickets, and Brahminy blind snakes. Some non-aging species, while rare, include sponges, planaria, quahog clams, Greenland sharks, some turtles, Aspen trees, and rockfish. - Recent Emergence: MAO-B’s harmful late-life allele may be evolutionarily young, not yet “weeded out”.
- Unknown Minor Benefits: Subtle advantages in youth remain undiscovered (though evidence is scant).
- Higher Level Selection: Older individuals are removed for population benefit.
The existence of a pure death gene (MAO-B) indicates that classical evolutionary theory may need refinement, or that we must consider newly emerging, selected-for “programmed aging” concepts while skeptically viewing the alternative of unselected, accidentally evolved “programmatic aging” being a random artifact of developmental processes becoming detrimental at older ages as being proposed by some (51).
Therapeutic Prospects
- MAO Inhibitors:
Selegiline/Deprenyl (MAO-B selective inhibitors) already show benefits in Parkinson’s and aging animal models (52). Could they be anti-aging therapies more broadly? - FAD Supplementation:
Directly boosting riboflavin or FAD might offset the MAO-driven deficit (53). - CD38 + MAO Dual-Targeting:
Restoring both NAD+ and FAD simultaneously could be a game-changing approach to mitigate the 80% combined ETC collapse. - MAO-A vs. MAO-B:
Inhibiting MAO-B specifically may yield fewer adverse effects than inhibiting MAO-A, given MAO-A’s essential roles in mood and aggression control (54).
Studies by Dr. József Knoll and others have reported that when Deprenyl (selegiline) is administered at higher doses—enough to broadly inhibit both MAO-B and MAO-A—it can extend the lifespans of laboratory animals by up to 40%. In rats, Knoll documented significant increases in average and maximal lifespan (particularly in older cohorts), linking these benefits to the compound’s broader anti‐oxidative and pro‐dopaminergic effects (55). At lower doses (5 mg/day in humans) Deprenyl irreversibly inhibits MAO-B and has a half-life of 40 days while at higher doses (20 mg/day) Deprenyl also inhibits MAO-A and for that reason has also been used as an anti-depressant (56). Parallel observations in beagles showed that Deprenyl administered at high doses not only enhanced longevity but also improved or sustained cognitive performance in aging dogs (57). Although the precise optimal dosage varies by species (e.g., around 40 mg/day for larger canines), the general consensus is that higher-dose Deprenyl’s ability to suppress both MAO isoforms preserves mitochondrial cofactors (like FAD) and reduces oxidative stress, delaying diverse degenerative processes of aging (58).
Areas for future research:
Potential “Death miRNAs”
Recent studies suggest that miR-128 may exemplify a “pure death miRNA,” accelerating age-related pathologies without providing significant early-life benefits. In multiple knockout (KO) models, loss of miR-128 appears to leave normal developmental processes intact while conferring resistance to degenerative cardiac, neurological, or metabolic decline (59). This fits the profile of a “true death gene” that, like certain death genes in other families (e.g., MAO-B), exerts few vital roles before orchestrating late-life deterioration. Though most miRNAs fulfill dual or pleiotropic functions across the lifespan, miR-128 stands out for its apparently negligible KO phenotype in early life. Ongoing research implicates additional candidates—such as miR-34 and miR-29—but these often maintain at least some regulatory or protective roles in development (60) (61). Consequently, miR-128 remains the leading example of a miRNA seemingly dedicated to aging-associated damage, thus meriting further evaluation as a “death miRNA” whose targeted inhibition may extend healthy lifespan.
The author has also hypothesized that LARP-1, which was discovered as the most activated gene in all tissues during mammalian aging in all mammals may also qualify as a true death gene (62). It is hypothesized that the version of LARP1 that causes aging is not the canonical, well-studied long LARP1 isoform which is a protein active in the cytoplasm at all ages. However, the author believes that the virtually unstudied short LARP1 isoform which codes for a lncRNA has only been seen active within the nucleus during embryogenesis (63) will also be found to active in aging/senile individuals in the nucleus as well. The purpose of short LARP1, the author believes, is to interfere with the RNA splicing process to cause truncated RNA or impaired protein RNA (introns not removed) to be produced during aging. Evidence for this is suggested by an unidentified isoform of LARP1 being found in the Harvard spliceosome database derived from Hela cells which has recently been removed for unknown reasons after the database managers were notified about it. The Horvath lab found that long LARP1 in the cytoplasm does not increase with aging even though LARP1 has been identified as the most important aging related gene that is activated during mammalian aging (personal communication). This is an excellent area for further investigation.
Conclusions
MAO-A and MAO-B both rise with age across multiple tissues, depleting FAD and impairing mitochondrial respiration. MAO-A retains partial beneficial roles (thus is only a “partial death gene”), whereas MAO-B stands out as the first confirmed instance of a true death gene: it confers negligible early-life benefit, yet surges in old age to undermine fundamental bioenergetics—mirroring the way CD38 escalates to deplete NAD+. This synergy of NAD+ and FAD depletion can drive a significant fraction of age-related disease phenotypes, from neurodegeneration to heart failure and cancer.
Future research should focus on definitive measurements of FAD depletion by MAO isoforms, the potential synergy of inhibiting both MAOs and CD38, and the evolutionary genetics behind a purely deleterious late-life gene. These insights challenge traditional evolutionary views, while also offering promising new therapeutic interventions to slow or even reverse aspects of programmed aging.
References
- Why do we age? Kirkwood, T. B., & Austad, S. N. . ,. 6809, (2000), Nature, Vol. 408, pp. 233-238.
- Horizons in the evolution of aging. Flatt, T., & Partridge, L. . (2018), BMC Biology,, Vol. 16, p. Artilce 93.
- Universal DNA methylation age across mammalian tissues. Lu, A.T., Fei, Z., Haghani, A., Horvath, S, et al. . (2023), Nat Aging, pp. 1144–1166 .
- Relation of Sex and Aging to Monoamine Oxidase Activity of Human Brain, Plasma, and Platelets. . Robinson, D. S., Davis, J. M., Nies, A., Ravaris, C. L., & Sylwester, D. . 6, (1971), Archives of General Psychiatry, , Vol. 24, pp. 536-539.
- Differential age-related changes of MAO-A and MAO-B in mouse brain and peripheral organs. Saura, J., Richards, J. G., & Mahy, N. 4, (1994)., Neurobiology of Aging, Vol. 15, pp. 399-408.
- Hormones for Perimenopausal and Postmenopausal Depression. . Hendrick, V. 1, Hendrick, V. (2004). , Psychiatric Times, , Vol. 21.
- Long-term administration of monoamine oxidase inhibitors alters the firing rate and pattern of dopamine neurons in the ventral tegmental area. Chenu, F., El Mansari, M., & Blier, P. 4, (2009)., International Journal of Neuropsychopharmacology, Vol. 12, pp. 475-485.
- Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. . Cases, O., Seif, I., Grimsby, J., Gaspar, P., Chen, K., Pournin, S., Müller, U., Aguet, M., Babinet, C., Shih, J. C., & De Maeyer, E. 5218, (1995)., Science, Vol. 268, pp. 1763-1766.
- Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. . Brunner, H. G., Nelen, M., Breakefield, X. O., Ropers, H. H., & van Oost, B. A. 5133, (1993)., Science,, Vol. 262, pp. 578-580.
- Monoamine oxidase: from genes to behavior. . Shih, J. C., Chen, K., & Ridd, M. J. 1, (1999)., Annual Review of Neuroscience,, Vol. 22, pp. 197-217.
- Monoamine oxidase inactivation: from pathophysiology to therapeutics. Bortolato, M., Chen, K., & Shih, J. C. (13-14), (2008). , Advanced Drug Delivery Reviews, Vol. 60, pp. 1527-1533.
- Social deficits and perseverative behaviors, but not overt aggression, in MAO-A hypomorphic mice. Bortolato, M., Chen, K., Godar, S. C., Chen, G., Wu, W., Rebrin, I., Farrell, M. R., Scott, A. L., Wellman, C. L., & Shih, J. C. 13, (2011)., Neuropsychopharmacology,, Vol. 36, pp. 2674-2688.
- Monoamine oxidase: from genes to behavior. . Shih, J. C., Chen, K., & Ridd, M. J. 1, 1999, Annual Review of Neuroscience, Vol. 22, pp. 197-217.
- Monoamine oxidase B prompts mitochondrial and cardiac dysfunction in pressure overloaded hearts. . Kaludercic, N., Carpi, A., Nagayama, T., Sivakumaran, V., Zhu, G., Lai, E. W., Bedja, D., De Mario, A., Chen, K., Gabrielson, K. L., Lindsey, M. L., Pacak, K., Takimoto, E., Shih, J. C., Kass, D. A., Di Lisa, F., & Paolocci, N. 2, (2014)., Antioxidants & Redox Signaling, , Vol. 20, pp. 267-280.
- Wang, X., … Pellegrini, M., Yang, L. Targeting monoamine oxidase A for T cell-based cancer immunotherapy. Wang, X., … Pellegrini, M., Yang, LSci. Immunol. 6(59):eabh2383, 2021.
- Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Youdim, M. B., Kupershmidt, L., Amit, T., & Weinreb, O. 2014, Parkinsonism & Related Disorders, Vol. 20, pp. S132-S136.
- CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Camacho-Pereira, J., Tarragó, M. G., Chini, C. C., Nin, V., Escande, C., Warner, G. M., … & Chini, E. N. 6, 2016, Cell metabolism, Vol. 23, pp. 1127-1139.
- The therapeutic potential of monoamine oxidase inhibitors. . Youdim, M. B., Edmondson, D., & Tipton, K. F. 4, 2006, Nature reviews neuroscience, Vol. 7, pp. 295-309.
- The CD38/NAD/SIRTUIN1/EZH2 axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. . Katsuyama, E., Suarez-Fueyo, A., Bradley, S. J., Mizui, M., Marin, A. V., Mulki, L., … & Tsokos, G. C. (2020). , Cell reports, p. 30.
- p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Villeneuve, C., Guilbeau-Frugier, C., Sicard, P., Lairez, O., Ordener, C., Duparc, T., … & Parini, A. 1, . (2013)., Antioxidants & redox signaling, Vol. 18, pp. 5-18.
- Oxidative stress by monoamine oxidase-A impairs transcription factor EB activation and autophagosome clearance, leading to cardiomyocyte necrosis and heart failure. Santin, Y., Sicard, P., Vigneron, F., Guilbeau-Frugier, C., Dutaur, M., Lairez, O., … & Mialet-Perez, J. 1, (2016). , Antioxidants & redox signaling, Vol. 25, pp. 10-27.
- Greater monoamine oxidase A binding in perimenopausal age as measured with carbon 11–labeled harmine positron emission tomography. Rekkas, P. V., Wilson, A. A., Lee, V. W., Yogalingam, P., Sacher, J., Rusjan, P., … & Meyer, J. H. 3, (2014)., JAMA psychiatry, Vol. 71, pp. 873-879.
- CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Camacho-Pereira, J., Tarragó, M. G., Chini, C. C., Nin, V., Escande, C., Warner, G. M., … & Chini, E. N. (201. 6, 016, Cell metabolism, Vol. 23, pp. 1127-1139.
- Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. . Blacher, E., Dadali, T., Bespalko, A., Haupenthal, V. J., Grimm, M. O., Hartmann, T., … & Levy, A. 1, (2015), Annals of neurology, Vol. 78, pp. 88-103.
- The therapeutic potential of monoamine oxidase inhibitors. . Youdim, M. B., Edmondson, D., & Tipton, K. F. 4, (2006), Nature reviews neuroscience, Vol. 7, pp. 295-309.
- Villeneuve, C., Guilbeau-Frugier, C., Sicard, P., Lairez, O., Ordener, C., Duparc, T., … & Parini, A. (2013). Villeneuve, C., Guilbeau-Frugier, C., Sicard, P., Lairez, O., Ordener, C., Duparc, T., … & Parini, A. 1, (2013), Antioxidants & redox signaling, Vol. 18, pp. 5-18.
- MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. . Mallajosyula, J. K., Kaur, D., Chinta, S. J., Rajagopalan, S., Rane, A., Nicholls, D. G., … & Andersen, J. K. 2, (2008). , PloS one, 3(2), e1616, Vol. 3, p. e1616.
- Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Schedin-Weiss, S., Inoue, M., Hromadkova, L., Teranishi, Y., Yamamoto, N. G., Wiehager, B., … & Tjernberg, L. O. 1, (2017). , Alzheimer’s research & therapy,, Vol. 9, pp. 1-19.
- Periodontitis: a potential risk factor for Alzheimer’s disease. . Cerajewska, T. L., Davies, M., & West, N. X. 1, (2015)., British dental journal, Vol. 218, pp. 29-34.
- CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. . Camacho-Pereira, J., Tarragó, M. G., Chini, C. C., Nin, V., Escande, C., Warner, G. M., … & Chini, E. N. 6, (2016), Cell metabolism, Vol. 23, pp. 1127-1139.
- Benefits in cardiac function by CD38 suppression. Guan, X. H., Hong, X., Zhao, N., Liu, X. H., Xiao, Y. F., Chen, T. T., … & Zhang, X. H. 2, (2022), American Journal of Physiology-Heart and Circulatory Physiology, Vol. 322, pp. H231-H243.
- Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Manzella, N., Santin, Y., Maggiorani, D., Martini, H., Douin-Echinard, V., Passos, J. F., … & Mialet-Perez, J. 5, (2018)., Aging cell, Vol. 17, p. e12811.
- p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. . Villeneuve, C., Guilbeau-Frugier, C., Sicard, P., Lairez, O., Ordener, C., Duparc, T., … & Parini, A. 1, (2013)., Antioxidants & redox signaling, Vol. 18, pp. 5-18.
- Monoamine oxidases, oxidative stress, and altered mitochondrial dynamics in cardiac ageing. Oxidative medicine and cellular longevity, 2017. Maggiorani, D., Manzella, N., Edmondson, D. E., Mattevi, A., Parini, A., Binda, C., & Mialet-Perez, J. 2017, Oxidative medicine and cellular longevity, .
- CD38 and CD157: a long journey from activation markers to multifunctional molecules. . Quarona, V., Zaccarello, G., Chillemi, A., Brunetti, E., Singh, V. K., Ferrero, E., … & Malavasi, F. 4, 2013, Cytometry Part B: Clinical Cytometry, Vol. 84, pp. 207-217.
- CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. . Chen, L., Diao, L., Yang, Y., Yi, X., Rodriguez, B. L., Li, Y., … & Wistuba, I. I. 5, (2018), Cancer discovery, Vol. 8, pp. 1156-1175.
- Monoamine oxidase A mediates prostate tumorigenesis and cancer metastasis. . Wu, J. B., Shao, C., Li, X., Li, Q., Hu, P., Shi, C., … & Zhau, H. E. 7, (2014), The Journal of clinical investigation, Vol. 124, pp. 2891-2908.
- MAOA-dependent activation of Shh-IL6-RANKL signaling network promotes prostate cancer metastasis by engaging tumor-stromal cell interactions. Wu, J. B., Yin, L., Shi, C., Li, Q., Duan, P., Huang, J. M., … & Zhau, H. E. 3, (2017), Cancer cell, Vol. 31, pp. 368-38.
- Stromal-derived MAOB promotes prostate cancer growth and progression through CXCL12-CXCR4 signaling. Xu, Y., Liu, H., Chen, J., & Zhou, Q. (. 6, 2024, Science Advances, Vol. 10, p. eadg3714.
- Monoamine oxidase B levels are highly expressed in human gliomas and are correlated with the expression of HiF-1α and with transcription factors Sp1 and Sp3. . Sharpe, M. A., & Baskin, D. S. 3, (2016), Oncotarget, Vol. 7, p. 3379.
- Cancer as a mitochondrial metabolic disease. . Seyfried, T. N. 43, (2015), Frontiers in Cell and Developmental Biology, Vol. 3.
- CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell metabolism, 23(6), 1127-1139. Camacho-Pereira, J., Tarragó, M. G., Chini, C. C., Nin, V., Escande, C., Warner, G. M., … & Chini, E. N. 6, 2016, Cell metabolism, Vol. 23, pp. 1127-1139.
- The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. Barbosa, M. T., Soares, S. M., Novak, C. M., Sinclair, D., Levine, J. A., Aksoy, P., & Chini, E. N. 13, 2007, The FASEB Journal, Vol. 21, pp. 3629-3639.
- Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. . Manzella, N., Santin, Y., Maggiorani, D., Martini, H., Douin-Echinard, V., Passos, J. F., … & Mialet-Perez, J. 5, 2018, Aging cel, Vol. 17, p. e12811.
- Obesity and insulin resistance are associated with lower monoamine oxidase B activity in blood. Mayoral-Mariles, A., Cruz-Martín del Campo, S. E., Diaz-Guerrero, M., Manjarrez-Gutierrez, G., Hernandez-Rodriguez, J., & Zenteno, E. 5, (2020), Neurochemical research, Vol. 45, pp. 1045-1052.
- Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. . Manzella, N., Santin, Y., Maggiorani, D., Martini, H., Douin-Echinard, V., Passos, J. F., … & Mialet-Perez, J. 5, 2018, Aging cell, Vol. 17, p. e12811.
- CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Camacho-Pereira, J., Tarragó, M. G., Chini, C. C., Nin, V., Escande, C., Warner, G. M., … & Chini, E. N. 6, (2016), Cell metabolism, Vol. 23, pp. 1127-1139.
- . NAD and the aging process: Role in life, death and everything in between. . Chini, C. C., Tarragó, M. G., & Chini, E. N. (2017), Molecular and cellular endocrinology, Vol. 455, pp. 62-74.
- p53-PGC-1α pathway mediates oxidative mitochondrial damage and cardiomyocyte necrosis induced by monoamine oxidase-A upregulation: role in chronic left ventricular dysfunction in mice. Antioxidants & redox signaling, 18(1), 5-18. Villeneuve, C., Guilbeau-Frugier, C., Sicard, P., Lairez, O., Ordener, C., Duparc, T., … & Parini, A. 1, (2013)., Antioxidants & redox signaling, Vol. 18, pp. 5-18.
- Lipopolysaccharide-induced epithelial monoamine oxidase mediates alveolar bone loss in a rat chronic wound model. Ekuni, D., Firth, J. D., Nayer, T., Tomofuji, T., Sanbe, T., Irie, K., … & Putnins, E. E. 4, (2009), The American journal of pathology,, Vol. 175.
- Genomes optimize reproduction: aging as a consequence of the developmental program. de Magalhães, J. P., & Church, G. M. 4, (2005), Physiology, Vol. 20, pp. 252-259.
- Deprenyl/selegiline after 50 years in research and therapy (1965-2015). Knoll, J. 9, 2016, Journal of Neural Transmission, Vol. 125, pp. 1003-1043.
- Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. Udhayabanu, T., Manole, A., Rajeshwari, M., Varalakshmi, P., Houlden, H., & Ashokkumar, B. 5, (2017), . Journal of Clinical Medicine, Vol. 6, p. 52.
- The therapeutic potential of monoamine oxidase inhibitors. . Youdim, M. B., Edmondson, D., & Tipton, K. F. 4, (2006), Nature reviews neuroscience, Vol. 7, pp. 295-309.
- Extension of life span of rats by long-term (-)deprenyl treatment. Knoll, J. 1, (1988), The Mount Sinai Journal of Medicine, Vol. 55, pp. 67-74.
- Evidence that formulations of the selective MAO-B inhibitor, selegiline, which bypass first-pass metabolism, also inhibit MAO-A in the human brain. . Fowler, J. S., Volkow, N. D., Logan, J., Alexoff, D. L., Iding, S. H., Wang, G. J., … & Cilento, R. 5, (2002), Neuropsychopharmacology, Vol. 27, pp. 867-876.
- Canine cognitive dysfunction as a model for human age-related cognitive decline, dementia and Alzheimer’s disease: clinical presentation, cognitive testing, pathology and response to 1-deprenyl therapy. . Ruehl, W. W., Bruyette, D. S., DePaoli, A., Cotman, C. W., Head, E., Milgram, N. W., & Cummings, B. J. . (1997), Progress in Brain Research, Vol. 115, pp. 283-293.
- Extension of life span of rats by long-term (-)deprenyl treatment. . Knoll, J. 1, (1988), The Mount Sinai Journal of Medicine,, Vol. 55, pp. 67-74.
- Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. . Huang, W., Feng, Y., Liang, J., Yu, H., Wang, C., Wang, B., Wang, M., Jiang, L., Meng, W., Cai, W., Medvedovic, M., Chen, J., Paul, C., Davidson, W. S., Sadayappan, S., Stambrook, P. J., Yu, X. Y., & Wang, Y. 1, 2018, Nature Communications, Vol. 9, p. 700.
- The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. . Liu, N., Landreh, M., Cao, K., Abe, M., Hendriks, G. J., Kennerdell, J. R., … & Bonini, N. M. 7386, (2012), Nature, Vol. 482, pp. 519-523.
- MicroRNA-29 is an essential regulator of brain maturation through regulation of CH methylation. . Swahari, V., Nakamura, A., Baran-Gale, J., Garcia, I., Crowther, A. J., Sons, R., … & Deshmukh, M. 1, (2021), Cell Reports, Vol. 35, p. 10894.
- Universal DNA methylation age across mammalian tissues. . Lu, A. T., Haghani, A., Li, C. Z., Lu, A. T., Robeck, T. R., Manley, N. B., … & Horvath, S. 9 1144-1166., s.l. : Nature Aging, 2023, Vol. 3.
- LARP1 isoform expression in human cancer cell lines. . Beggs, A.D., and Blagden, S.P. 2 237-247, s.l. : RNA Biology, 18(2), 237-247, (2021), Vol. 18.